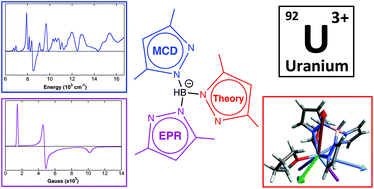
Homoleptic Uranium–Bis(acyl)phosphide Complexes
Anion-induced disproportionation of Th(III) complexes to form Th(II) and Th(IV) products
Side-on coordination of diphosphorus to a mononuclear iron center
A TMEDA–Iron Adduct Reaction Manifold in Iron-Catalyzed C(sp2)−C(sp3) Cross-Coupling Reactions
Stephanie H. Carpenter, Nikki J. Wolford, Brennan S. Billow, Taylor V. Fetrow, Nathalia Cajiao, Aleksa Radović, Michael T. Janicke, Michael L. Neidig, and Aaron M. Tondreau
Inorg. Chem. 2022
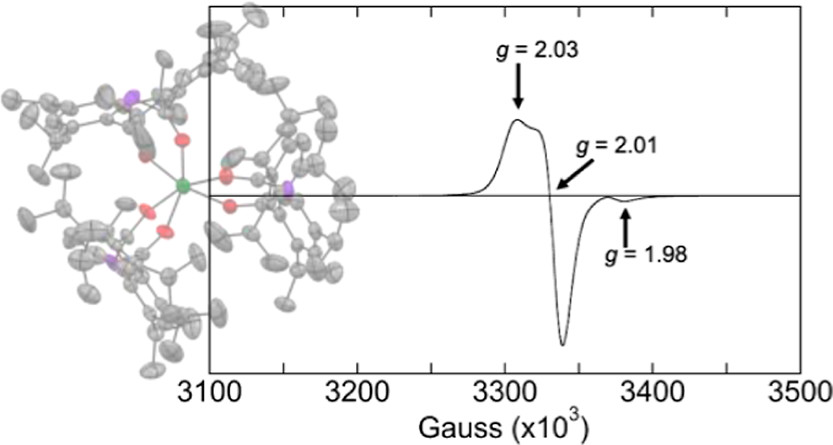
The first uranium bis(acyl)phosphide (BAP) complexes were synthesized from the reaction between sodium bis(mesitoyl)phosphide (Na(mesBAP)) or sodium bis(2,4,6-triisopropylbenzoyl)phosphide (Na(trippBAP)) and UI3(1,4-dioxane)1.5. Thermally stable, homoleptic BAP complexes were characterized by single-crystal X-ray diffraction and electron paramagnetic resonance (EPR) spectroscopy, when appropriate, for the elucidation of the electronic structure and bonding of these complexes. EPR spectroscopy revealed that the BAP ligands on the uranium center retain a significant amount of electron density. The EPR spectrum of the trivalent U(trippBAP)3 has a rhombic signal near g = 2 (g1 = 2.03; g2 = 2.01; and g3 = 1.98) that is consistent with the EPR-observed unpaired electron being located in a molecular orbital that appears ligand-derived. However, upon warming the complex to room temperature, no resonance was observed, indicating the presence of uranium character.
Justin C. Wedal, Nathalia Cajiao, Michael L. Neidig and William J. Evans
Chem. Commun. 2022, 58, 5289-5291
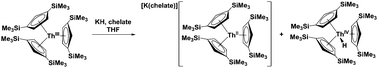
A new synthesis of Th(II) complexes has been identified involving addition of simple MX salts (M = Li, Na, K; X = H, Cl, Me, N3) to Cp′′3ThIII [Cp′′ = [C5H3(SiMe3)2] in the presence of 18-crown-6 or 2.2.2-cryptand, forming [M(chelate)][Cp′′3ThII] and Cp′′3ThIVX. Cptet3ThIII (Cptet = C5Me4H) reacts with KH to form Cptet3ThIVH and the C–H bond activation product, [K(crypt)]{[Cptet2ThIVH[η1:η5-C5Me3H(CH2)]}.
Shuai Wang, Jeffrey D. Sears, Curtis E. Moore, Arnold L. Rheingold, Michael L. Neidig, Joshua S. Figueroa
Science 2022, 375, 1393-1397
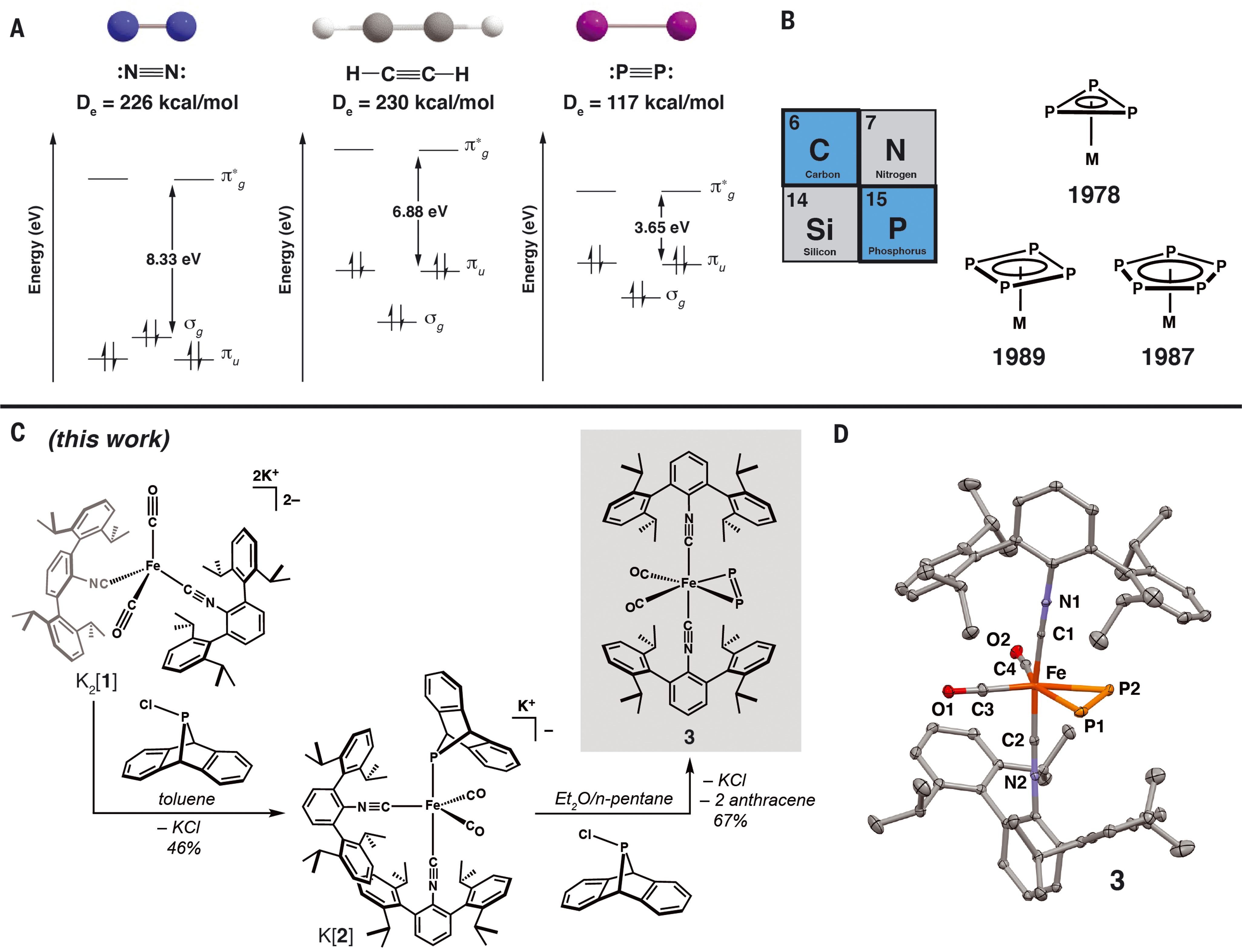
The diagonal relationship in the periodic table between phosphorus and carbon has set an expectation that the triple-bonded diatomic diphosphorus molecule (P2) should more closely mimic the attributes of acetylene (HC≡CH) rather than its group 15 congener dinitrogen (N2). Although acetylene has well-documented coordination chemistry with mononuclear transition metals, coordination complexes that feature P2 bound to a single metal center have remained elusive. We report the isolation and x-ray crystallographic characterization of a mononuclear iron complex featuring P2 coordination in a side-on, η2-binding mode. An analogous η2-bound bis-timethylsilylacetylene iron complex is reported for comparison. Nuclear magnetic resonance, infrared, and Mössbauer spectroscopic analysis—in conjunction with density functional theory calculations—demonstrate that η2-P2 and η2-acetylene ligands exert a similar electronic demand on mononuclear iron centers but exhibit different reactivity profiles.
Nikki J. Bakas, Dr. Jeffrey D. Sears, Dr. William W. Brennessel, Prof. Dr. Michael L. Neidig
Angew. Chem. Int. Ed. 2022, 61, e202114986
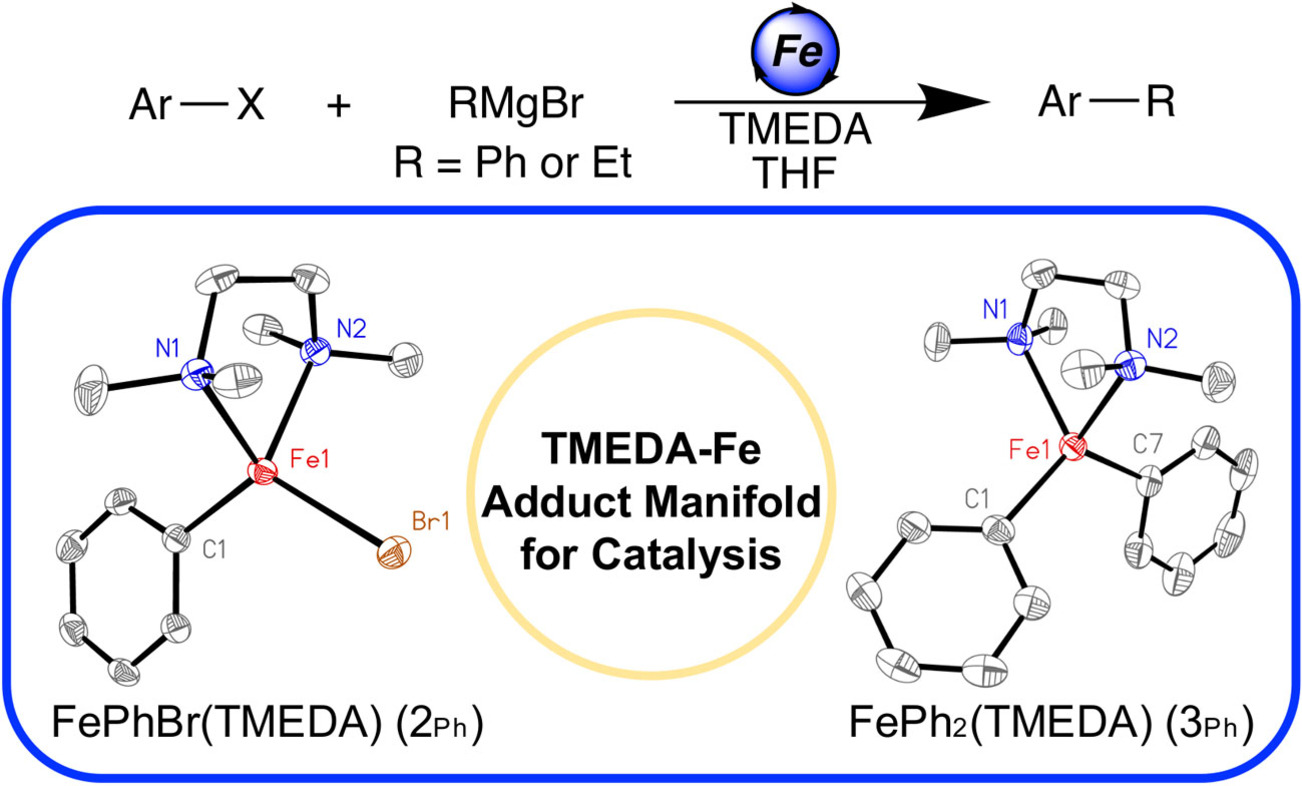
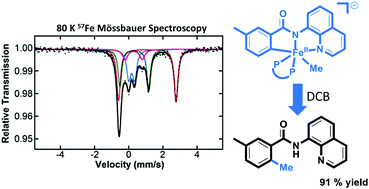
Historically, the role of TMEDA in iron-catalyzed C(sp2)−C(sp3) cross-coupling has been disputed due to a lack of insight into the iron species present during catalysis. In this report, low-temperature synthesis, freeze-trapped 57Fe Mössbauer, X-ray diffraction (XRD), and chemical-quenched GC analysis were utilized to identify highly reactive and selective TMEDA-ligated iron species responsible for C(sp2)−C(sp3) cross-coupling catalysis.
2021
Creation of an unexpected plane of enhanced covalency in cerium(III) and berkelium(III) terpyridyl complexes
Intermediates and mechanism in iron-catalyzed C–H methylation with trimethylaluminum
General method for iron-catalyzed multicomponent radical cascades–cross-couplings
Air-Stable Iron-Based Precatalysts for Suzuki–Miyaura Cross-Coupling Reactions between Alkyl Halides and Aryl Boronic Esters
An anionic iron-hydride superstar for the isomerization of terminal alkenes
Dilithium Amides as a Modular Bis-Anionic Ligand Platform for Iron-Catalyzed Cross-Coupling
NHC Effects on Reduction Dynamics in Iron-catalyzed Organic Transformations
Additive and Counterion Effects in Iron-Catalyzed Reactions Relevant to C–C Bond Formation
Experimental and computational studies of the mechanism of iron-catalysed C–H activation/functionalisation with allyl electrophiles
Recent Advances in Synthesis, Characterization and Reactivities of Iron-Alkyl and Iron-Aryl Complexes
Characterization Methods for Paramagnetic Organometallic Complexes
Iron-catalyzed C-H activation/functionalization to form C-C bonds
[2Fe–2S] Cluster Supported by Redox-Active o-Phenylenediamide Ligands and Its Application toward Dinitrogen Reduction
Near-infrared C-term MCD spectroscopy of octahedral uranium(v) complexes
Forged in iron
Activation of ammonia and hydrazine by electron rich Fe(II) complexes supported by a dianionic pentadentate ligand platform through a common terminal Fe(III) amido intermediate
C-H Activation/Functionalization With Earth Abundant 3d Transition Metals
Metal-Carbon Bonds of Iron and Manganese
C-Term magnetic circular dichroism (MCD) spectroscopy in paramagnetic transition metal and f-element organometallic chemistry
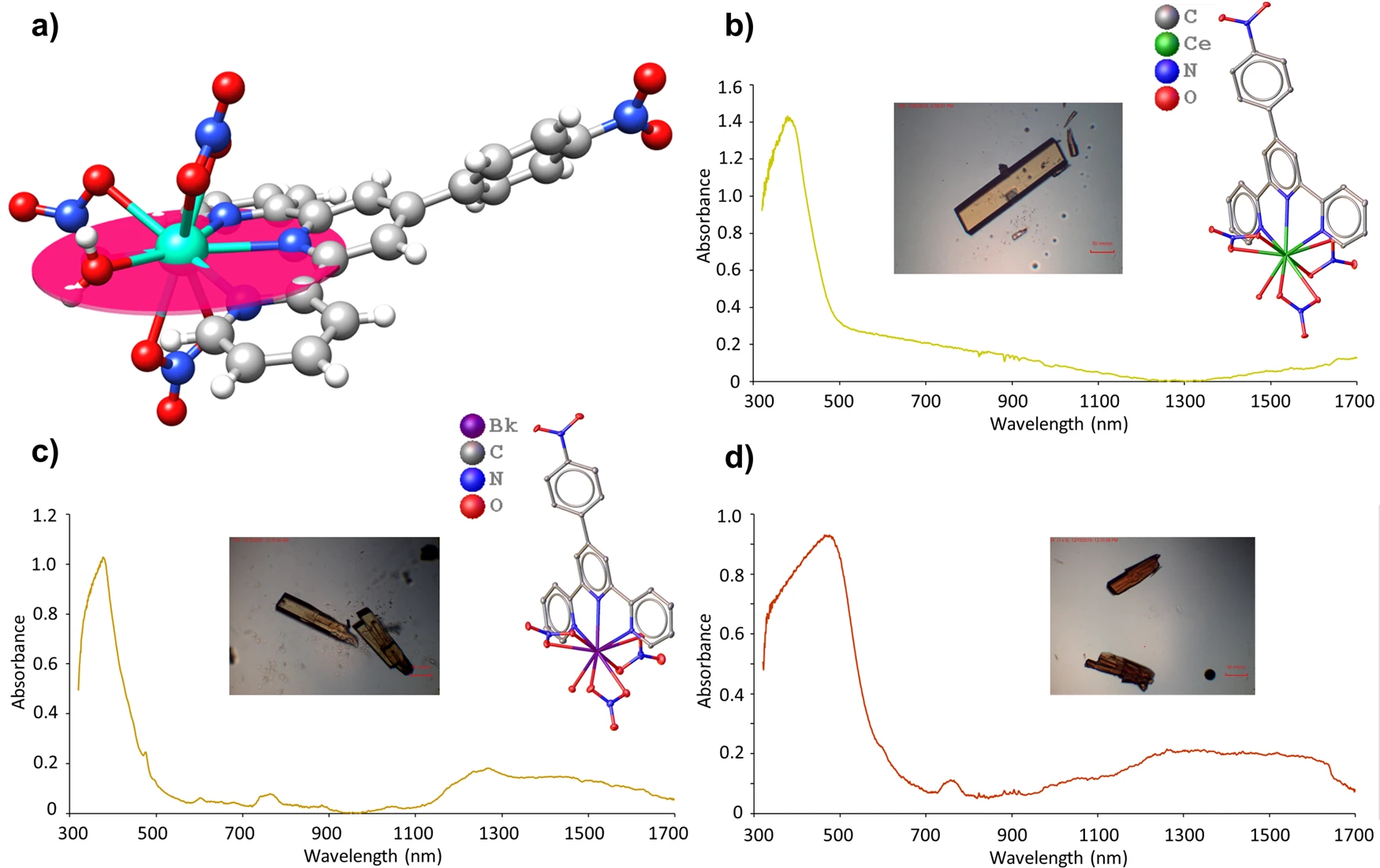
Alyssa N. Gaiser, Cristian Celis-Barros, Frankie D. White, Maria J. Beltran-Leiva, Joseph M. Sperling, Sahan R. Salpage, Todd N. Poe, Daniela Gomez Martinez, Tian Jian, Nikki J. Wolford, Nathaniel J. Jones, Amanda J. Ritz, Robert A. Lazenby, John K. Gibson, Ryan E. Baumbach, Dayán Páez-Hernández, Michael L. Neidig, Thomas E. Albrecht-Schönzart
Nat. Commun. 2021, 12, 7230
Controlling the properties of heavy element complexes, such as those containing berkelium, is challenging because relativistic effects, spin-orbit and ligand-field splitting, and complex metal-ligand bonding, all dictate the final electronic states of the molecules. While the first two of these are currently beyond experimental control, covalent M‒L interactions could theoretically be boosted through the employment of chelators with large polarizabilities that substantially shift the electron density in the molecules. This theory is tested by ligating BkIII with 4’-(4-nitrophenyl)-2,2’:6’,2”-terpyridine (terpy*), a ligand with a large dipole. The resultant complex, Bk(terpy*)(NO3)3(H2O)·THF, is benchmarked with its closest electrochemical analog, Ce(terpy*)(NO3)3(H2O)·THF. Here, we show that enhanced Bk‒N interactions with terpy* are observed as predicted. Unexpectedly, induced polarization by terpy* also creates a plane in the molecules wherein the M‒L bonds trans to terpy* are shorter than anticipated. Moreover, these molecules are highly anisotropic and rhombic EPR spectra for the CeIII complex are reported.
Shilpa Bhatia, Joshua C. DeMuth and Michael L. Neidig
Chem. Commun. 2021, 57, 12784-12787
A mechanistic study is performed on the reaction method for iron-catalyzed C–H methylation with AlMe3 reagent, previously proposed to involve cyclometalated iron(III) intermediates and an iron(III)/(I) reaction cycle. Detailed spectroscopic studies (57Fe Mössbauer, EPR) during catalysis and in stoichiometric reactions identify iron(II) complexes, including cyclometalated iron(II) intermediates, as the major iron species formed in situ under catalytic reaction conditions. Reaction studies identify a cyclometalated iron(II)-methyl species as the key intermediate leading to C–H methylated product upon reaction with oxidant, consistent with a previously proposed iron(II)/iron(III)/iron(I) reaction manifold for C–H arylation.
Lei Liu, Maria Camila Aguilera, Wes Lee, Cassandra R. Youshaw, Michael L. Neidig, And Osvaldo Gutierrez
Science 2021, 374, 432-439
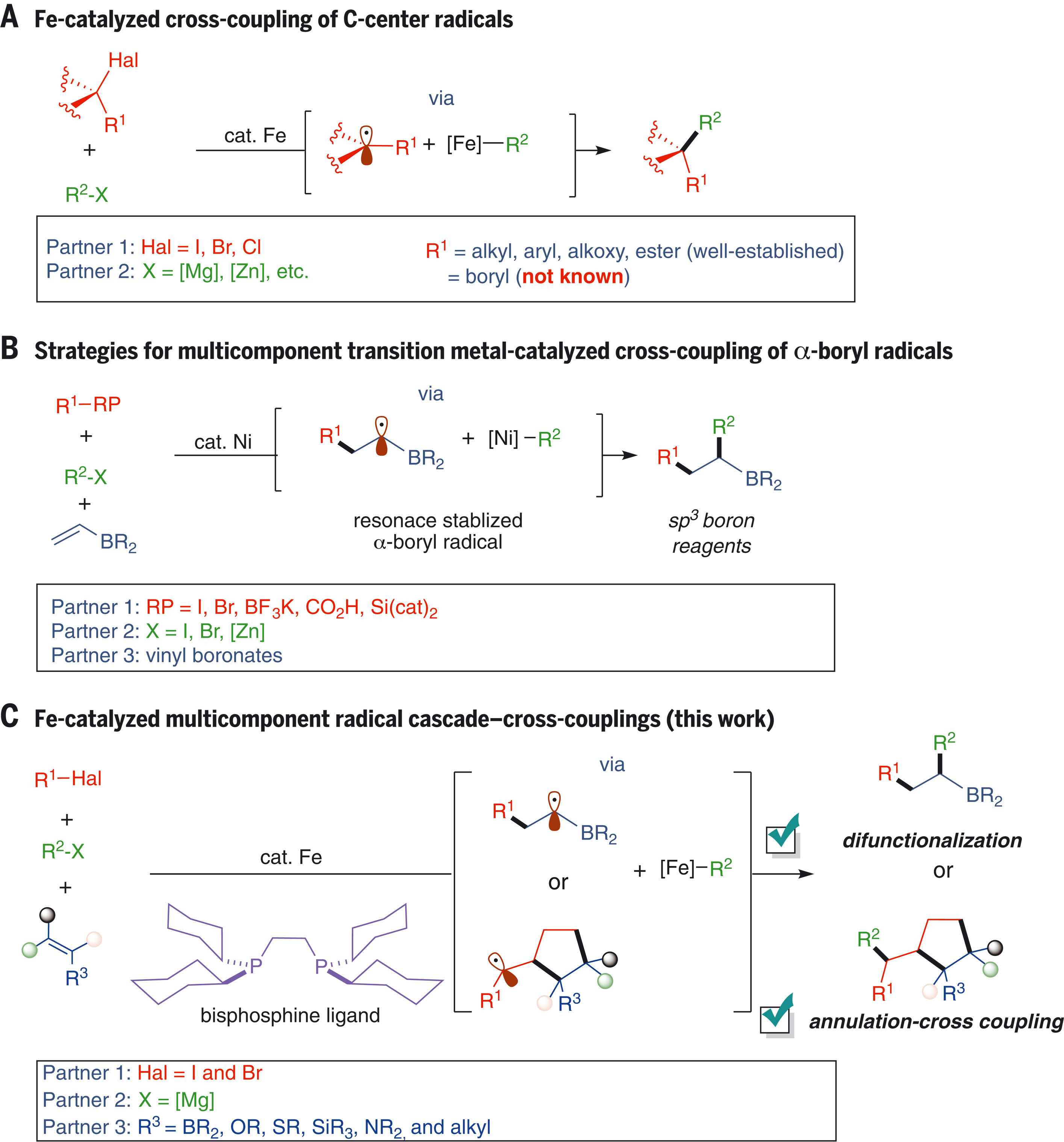
Transition metal–catalyzed cross-coupling reactions are some of the most widely used methods in chemical synthesis. However, despite notable advantages of iron (Fe) as a potentially cheaper, more abundant, and less toxic transition metal catalyst, its practical application in multicomponent cross-couplings remains largely unsuccessful. We demonstrate 1,2-bis(dicyclohexylphosphino)ethane Fe–catalyzed coupling of α-boryl radicals (generated from selective radical addition to vinyl boronates) with Grignard reagents. Then, we extended the scope of these radical cascades by developing a general and broadly applicable Fe-catalyzed multicomponent annulation–cross-coupling protocol that engages a wide range of π-systems and permits the practical synthesis of cyclic fluorous compounds. Mechanistic studies are consistent with a bisarylated Fe(II) species being responsible for alkyl radical generation to initiate catalysis, while carbon-carbon bond formation proceeds between a monoarylated Fe(II) center and a transient alkyl radical.
Alexander S. Wong, Bufan Zhang, Bo Li, Michael L. Neidig, and Jeffery A. Byers
Org. Process Res. Dev. 2021
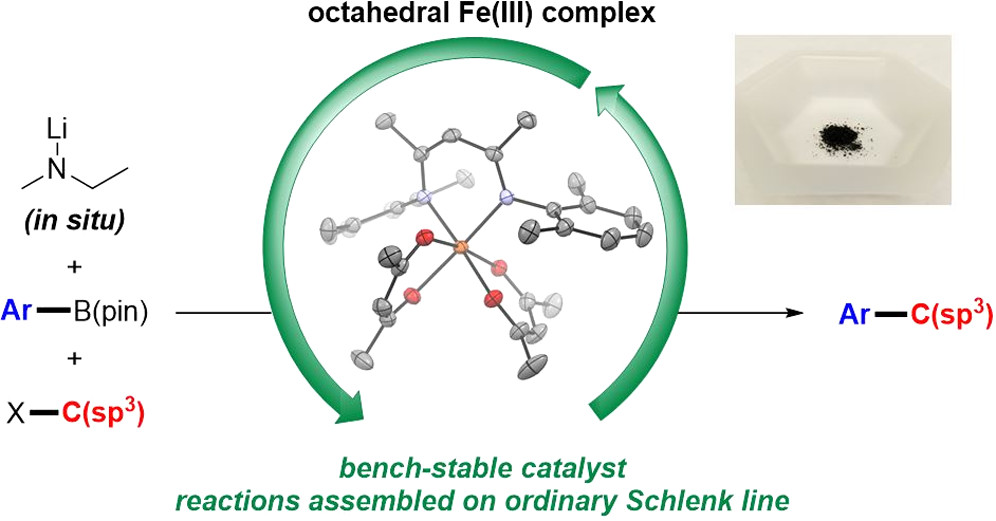
The development of an air-stable iron(III)-based precatalyst for the Suzuki–Miyaura cross-coupling reaction of alkyl halides and unactivated aryl boronic esters is reported. Despite benefits to cost and toxicity, the proclivity of iron(II)-based complexes to undergo deactivation via oxidation or hydrolysis is a limiting factor for their widespread use in cross-coupling reactions compared to palladium-based or nickel-based complexes. The new octahedral iron(III) complex demonstrates long-term stability on the benchtop as assessed by a combination of 1H NMR spectroscopy, Mössbauer spectroscopy, and its sustained catalytic activity after exposure to air. The improved stability of the iron-based catalyst facilitates an improved protocol in which Suzuki–Miyaura cross-coupling reactions of valuable substrates can be assembled without the use of a glovebox and access a diverse scope of products similar to reactions assembled in the glovebox with iron(II)-based catalysts.
Maria Camila Aguilera, Michael L. Neidig
Chem Catalysis 2021, 1, 488-489
In this issue of Chem Catalysis, de Ruiter and co-workers report a highly efficient and selective method for isomerization of a wide variety of terminal alkenes, employing the well-defined anionic complex [(PCNHCP)Fe(H)N2][Na]. This work also includes important insights about the structure and reactivity of the iron intermediates involved in the catalytic cycle.
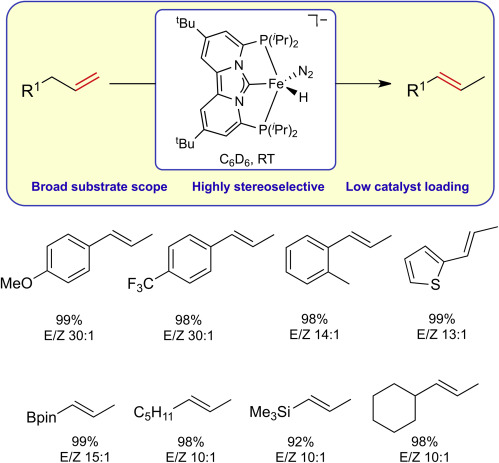
Peter G. N. Neate, Bufan Zhang, Jessica Conforti, William W. Brennessel, and Michael L. Neidig
Org. Lett. 2021, 23, 5958-5963
Dilithium amides have been developed as a bespoke and general ligand for iron-catalyzed Kumada–Tamao–Corriu cross-coupling reactions, their design taking inspiration from previous mechanistic and structural studies. They allow for the cross-coupling of alkyl Grignard reagents with sp2-hybridized electrophiles as well as aryl Grignard reagents with sp3-hybridized electrophiles. This represents a rare example of a single iron-catalyzed system effective across diverse coupling reactions without significant modification of the catalytic protocol, as well as remaining operationally simple.
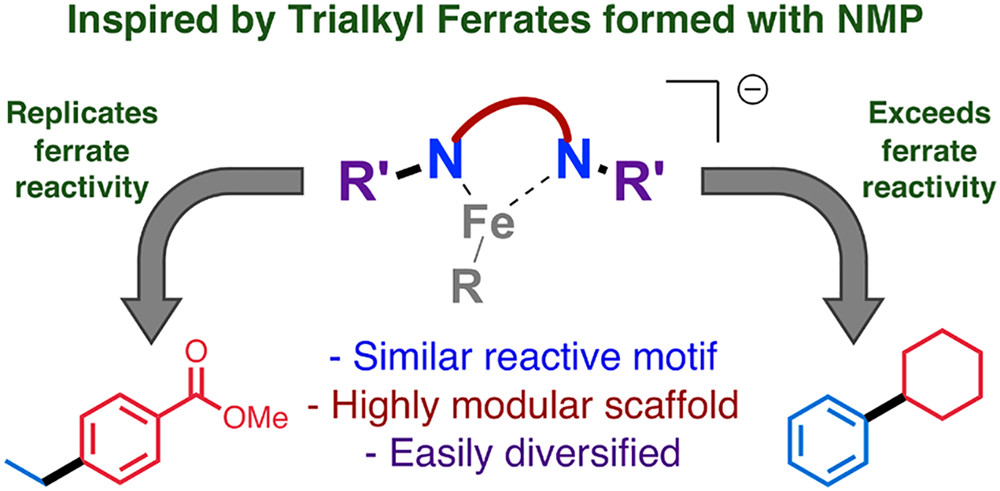
Nikki J Wolford, Salvador B Munoz III, Peter G. N. Neate, William W. Brennessel, Michael Neidig
Chem. Eur. J. 2021, 27, 13651-13658
The high abundance, low toxicity and rich redox chemistry of iron has resulted in a surge of iron catalyzed organic transformations over the last two decades. Within this area, N -heterocyclic carbene (NHC) ligands have been widely utilized to achieve high yields across reactions including cross-coupling and C-H alkylation, amongst others. Central to the development of iron-NHC catalytic methods is the understanding of iron speciation and the propensity of these species to undergo reduction events, as low-valent iron species can be advantageous or undesirable from one system to the next. This study highlights the importance of the identity of the NHC on iron speciation upon reaction with EtMgBr, where reactions with SIMes and IMes NHCs were shown to undergo β-hydride elimination more readily than those with SIPr and IPr NHCs. This insight is vital to developing new iron-NHC catalyzed transformations as understanding how to control this reduction by simply changing the NHC is central to improving the reactivity in iron-NHC catalysis.
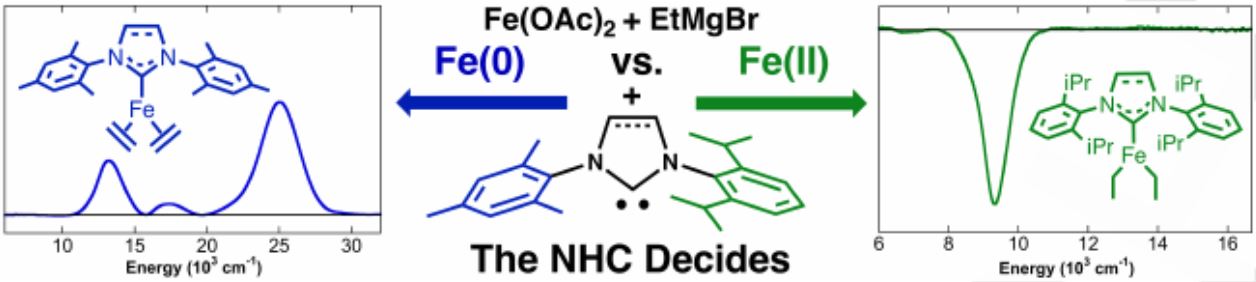
Nikki J. Bakas and Michael L. Neidig
ACS Catal. 2021, 11, 8493–8503
The use of iron catalysts in carbon–carbon bond forming reactions is of interest as an alternative to precious metal catalysts, offering reduced cost, lower toxicity, and different reactivity. While well-defined ligands such as N-heterocyclic carbenes (NHCs) and phosphines can be highly effective in these reactions, additional additives such as N-methylpyrrolidone (NMP), N,N,N′,N′-tetramethylethylenediamine (TMEDA), and iron salts that alter speciation can also be employed to achieve high product yields. However, in contrast to well-defined iron ligands, the roles of these additives are often ambiguous, and molecular-level insights into how they achieve effective catalysis are not well-defined. Using a unique physical–inorganic in situ spectroscopic approach, detailed insights into the effect of additives on iron speciation, mechanism, and catalysis can inform further reaction development. In this Perspective, recent advances will be discussed as well as ongoing challenges and potential opportunities in iron-catalyzed reactions.
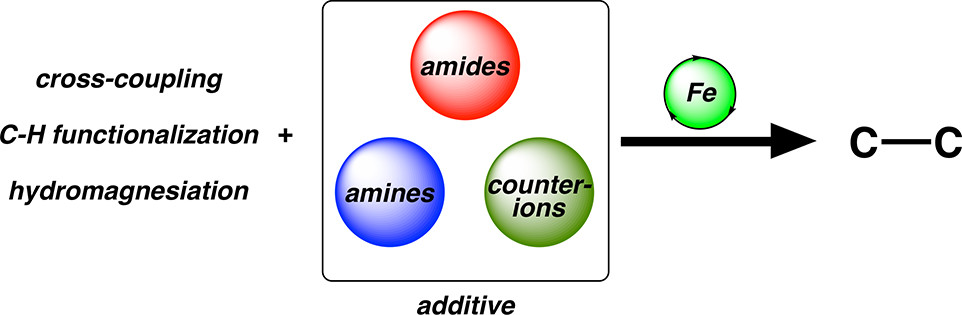
Joshua C. DeMuth, Zhihui Song, Stephanie H. Carpenter, Theresa E. Boddie, Aleksa Radović, Tessa M. Baker, Osvaldo Gutierrez and Michael L. Neidig
Chem. Sci. 2021, 12, 9398-9407
Synthetic methods that utilise iron to facilitate C–H bond activation to yield new C–C and C–heteroatom bonds continue to attract significant interest. However, the development of these systems is still hampered by a limited molecular-level understanding of the key iron intermediates and reaction pathways that enable selective product formation. While recent studies have established the mechanism for iron-catalysed C–H arylation from aryl-nucleophiles, the underlying mechanistic pathway of iron-catalysed C–H activation/functionalisation systems which utilise electrophiles to establish C–C and C–heteroatom bonds has not been determined. The present study focuses on an iron-catalysed C–H allylation system, which utilises allyl chlorides as electrophiles to establish a C–allyl bond. Freeze-trapped inorganic spectroscopic methods (57Fe Mössbauer, EPR, and MCD) are combined with correlated reaction studies and kinetic analyses to reveal a unique and rapid reaction pathway by which the allyl electrophile reacts with a C–H activated iron intermediate. Supporting computational analysis defines this novel reaction coordinate as an inner-sphere radical process which features a partial iron–bisphosphine dissociation. Highlighting the role of the bisphosphine in this reaction pathway, a complementary study performed on the reaction of allyl electrophile with an analogous C–H activated intermediate bearing a more rigid bisphosphine ligand exhibits stifled yield and selectivity towards allylated product. An additional spectroscopic analysis of an iron-catalysed C–H amination system, which incorporates N-chloromorpholine as the C–N bond-forming electrophile, reveals a rapid reaction of electrophile with an analogous C–H activated iron intermediate consistent with the inner-sphere radical process defined for the C–H allylation system, demonstrating the prevalence of this novel reaction coordinate in this sub-class of iron-catalysed C–H functionalisation systems. Overall, these results provide a critical mechanistic foundation for the rational design and development of improved systems that are efficient, selective, and useful across a broad range of C–H functionalisations.
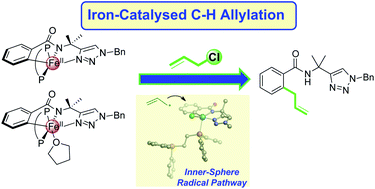
Bufan Zhang, Maria Camila Aguilera, Nathalia Cajiao, Michael L. Neidig
Comprehensive Organometallic Chemistry IV, K. Meyer, D. O’Hare, G. Parkin, Eds., Elsavier, 2021
The use of iron in catalysis continues to receive significant attention due to its high abundance, low toxicity, and unique reactivity. In numerous reactions, organoiron species, including iron-alkyl and iron-aryl complexes, have been identified as key reactive intermediates. Motivated by these roles and catalysis as well as broader interest into the fundamental chemistry of such complexes, extensive research has focused on synthesis, reactivity and electronic structure and bonding in iron-alkyl and iron-aryl complexes. This chapter highlights recent examples of iron-alkyl and iron-aryl complexes.
Aleksa Radović, Shilpa Bhatia, Michael L. Neidig
Comprehensive Organometallic Chemistry IV, K. Meyer, D. O’Hare, G. Parkin, Eds., Elsavier, 2021
As the use of base metals in organometallic chemistry continues to expand, the ability to characterize open shell organometallic compounds with unpaired electrons is essential. This chapter describes four advanced techniques that are particularly powerful for evaluating electronic structure, bonding and reactivity in paramagnetic organometallic complexes: electron paramagnetic resonance, magnetic circular dichroism, X-ray absorption and nuclear magnetic resonance spectroscopies. A discussion of the fundamentals of each spectroscopic method is presented to provide the necessary background and theory central to each technique. This is followed by discussions of key applications of each method for paramagnetic organometallic complexes, using specific examples from the literature to highlight the insight into electronic structure, bonding and reactivity that these methods can provide.
Michael L. Neidig, Shilpa Bhatia, Joshua C. DeMuth
Handbook of C-H Functionalization, 2021, in press
Qiuming Liang, Joshua C. DeMuth, Aleksa Radović, Nikki J. Wolford, Michael L. Neidig, and Datong Song
Inorg. Chem. 2021, 60, 13811-13820
As prevalent cofactors in living organisms, iron–sulfur clusters participate in not only the electron-transfer processes but also the biosynthesis of other cofactors. Many synthetic iron–sulfur clusters have been used in model studies, aiming to mimic their biological functions and to gain mechanistic insight into the related biological systems. The smallest [2Fe–2S] clusters are typically used for one-electron processes because of their limited capacity. Our group is interested in functionalizing small iron–sulfur clusters with redox-active ligands to enhance their electron storage capacity, because such functionalized clusters can potentially mediate multielectron chemical transformations. Herein we report the synthesis, structural characterization, and catalytic activity of a diferric [2Fe–2S] cluster functionalized with two o-phenylenediamide ligands. The electrochemical and chemical reductions of such a cluster revealed rich redox chemistry. The functionalized diferric cluster can store up to four electrons reversibly, where the first two reduction events are ligand-based and the remainder metal-based. The diferric [2Fe–2S] cluster displays catalytic activity toward silylation of dinitrogen, affording up to 88 equiv of the amine product per iron center.
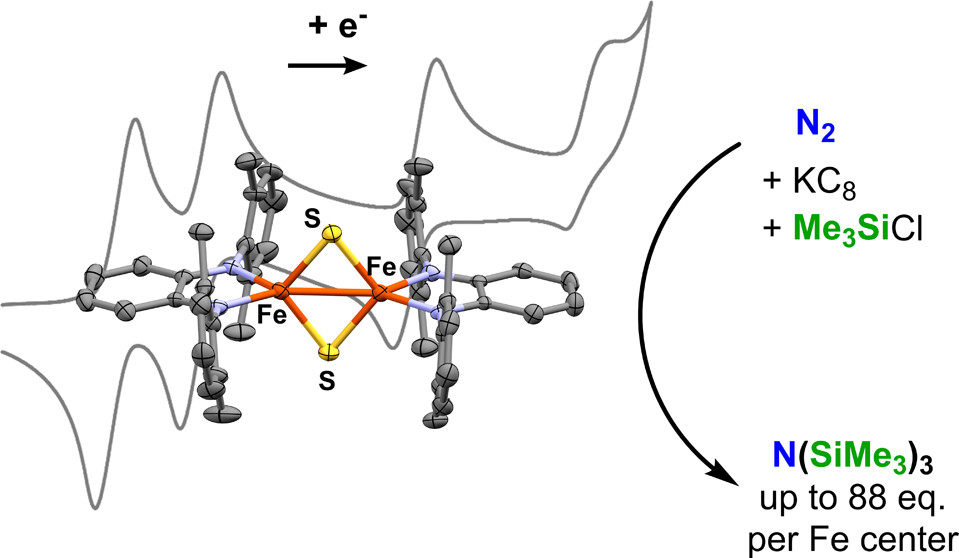
Daniel J. Curran, Gaurab Ganguly, Yonaton N. Heit, Nikki J. Wolford, Stefan G. Minasian, Matthias W. Löble, Samantha K. Cary, Stosh A. Kozimor, Jochen Autschbach and Michael L. Neidig
Dalton Trans. 2021, 50, 5483–5492
C-term magnetic circular dichroism (MCD) spectroscopy is a powerful method for probing d–d and f–f transitions in paramagnetic metal complexes. However, this technique remains underdeveloped both experimentally and theoretically for studies of U(V) complexes of Oh symmetry, which have been of longstanding interest for probing electronic structure, bonding, and covalency in 5f systems. In this study, C-term NIR MCD of the Laporte forbidden f–f transitions of [UCl6]− and [UF6]− are reported, demonstrating the significant fine structure resolution possible with this technique including for the low energy Γ7 → Γ8 transitions in [UF6]−. The experimental NIR MCD studies were further extended to [U(OC6F5)6]−, [U(CH2SiMe3)6]−, and [U(NC(tBu)(Ph))6]− to evaluate the effects of ligand-type on the f–f MCD fine structure features. Theoretical calculations were conducted to determine the Laporte forbidden f–f transitions and their MCD intensity experimentally observed in the NIR spectra of the U(V) hexahalide complexes, via the inclusion of vibronic coupling, to better understand the underlying spectral fine structure features for these complexes. These spectra and simulations provide an important platform for the application of MCD spectroscopy to this widely studied class of U(V) complexes and identify areas for continued theoretical development.
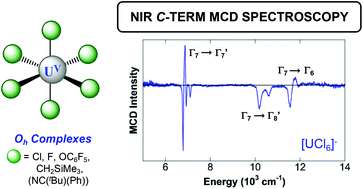
Peter G. N. Neate, Michael L. Neidig
Nat Rev Chem 2021, 5, 223–224
Fifty years ago, Kochi reported the iron-catalysed cross-coupling of alkenyl halides and alkyl Grignard reagents. Sparking a cross-coupling revolution, we reflect on the impact of this achievement and the importance of iron in the development of cross-coupling catalysis.
Lucie Nurdin, Yan Yang, Peter G. N. Neate, Warren E. Piers, Laurent Maron, Michael L. Neidig, Jian-Bin Lina and Benjamin S. Gelfand
Chem. Sci. 2021, 50, 416–428
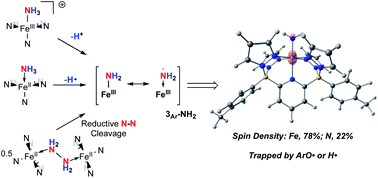
We report the use of electron rich iron complexes supported by a dianionic diborate pentadentate ligand system, B2Pz4Py, for the coordination and activation of ammonia (NH3) and hydrazine (NH2NH2). For ammonia, coordination to neutral (B2Pz4Py)Fe(II) or cationic [(B2Pz4Py)Fe(III)]+ platforms leads to well characterized ammine complexes from which hydrogen atoms or protons can be removed to generate, fleetingly, a proposed (B2Pz4Py)Fe(III)–NH2 complex (3Ar-NH2). DFT computations suggest a high degree of spin density on the amido ligand, giving it significant aminyl radical character. It rapidly traps the H atom abstracting agent 2,4,6-tri-tert-butylphenoxy radical (ArO˙) to form a C–N bond in a fully characterized product (2Ar), or scavenges hydrogen atoms to return to the ammonia complex (B2Pz4Py)Fe(II)–NH3 (1Ar-NH3). Interestingly, when (B2Pz4Py)Fe(II) is reacted with NH2NH2, a hydrazine bridged dimer, (B2Pz4Py)Fe(II)–NH2NH2–Fe(II)(B2Pz4Py) ((1Ar)2-NH2NH2), is observed at −78 °C and converts to a fully characterized bridging diazene complex, 4Ar, along with ammonia adduct 1Ar-NH3 as it is allowed to warm to room temperature. Experimental and computational evidence is presented to suggest that (B2Pz4Py)Fe(II) induces reductive cleavage of the N–N bond in hydrazine to produce the Fe(III)–NH2 complex 3Ar-NH2, which abstracts H˙ atoms from (1Ar)2-NH2NH2 to generate the observed products. All of these transformations are relevant to proposed steps in the ammonia oxidation reaction, an important process for the use of nitrogen-based fuels enabled by abundant first row transition metals.
Michael L. Neidig, Joshua C. DeMuth, Bufan Zhang
CComprehensive Coordination Chemistry III, Constable, E. C., Parkin, G., Que Jr, L., Eds., Elsavier, Vol 9, 2021, 260-310
The rapid development of C-H functionalization reactions can be attributed to their direct synthetic approach and attractive atom-economy. Precious metal catalysis has been extensively studied and proven effective. In recent years, 3d metals have become increasingly popular due to their high abundance, low toxicity and unique reactivity. This chapter provides an overview of 3d metal-catalyzed C-H functionalization reactions, featuring iron, cobalt, nickel and copper.

Michael L. Neidig, Nikki J. Bakas, Peter G.N. Neate, Jeffrey D. Sears
CComprehensive Coordination Chemistry III, Constable, E. C., Parkin, G., Que Jr, L., Eds., Elsavier, Vol 5, 2021, 82-122
This chapter encompasses organometallics complexes containing both iron- and manganese-carbon bond moieties. Due to the volume of literature the content of this chapter has been restricted to the exclusion of iron- and manganese-carbon bonded complexes containing only carbonyl (CO) and cyclopentadienide (Cp−) fragments. Instead, this chapter focuses on the use of additives such as phosphine, nitrogen, and carbene donor ligands which stabilize iron- and manganese-carbon bond moieties of selected organometallic complexes.
Nikki J. Wolford, Aleksa Radovic and Michael L. Neidig
Dalton Trans. 2021, 50, 416–428
Magnetic circular dichroism (MCD) spectroscopy is a powerful experiment used to probe the electronic structure and bonding in paramagnetic metal-based complexes. While C-term MCD spectroscopy has been utilized in many areas of chemistry, it has been underutilized in studying paramagnetic organometallic transition metal and f-element complexes. From the analysis of isolated organometallic complexes to the study of in situ generated species, MCD can provide information regarding ligand interactions, oxidation and spin state, and geometry and coordination environment of paramagnetic species. The pratical aspects of this technique, such as air-free sample preparation and cryogenic experimental temperatures, allow for the study of highly unstable species, something that is often difficult with other spectroscopic techniques. This perspective highlights MCD studies of both transition metal and f-element organometallic complexes, including in situ generated reactive intermediates, to demonstrate the utility of this technique in probing electronic structure, bonding and mechanism in paramagnetic organometallic chemistry.
2020
Open Shell Iron Catalysis: Mechanistic Challenges, Approaches and Pitfalls
Ligand effects on electronic structure and bonding in U(III) coordination complexes: a combined MCD, EPR and computational study
Identifying correlations in Fischer-Tropsch synthesis and CO2 hydrogenation over Fe-based ZSM-5 catalysts
Syntheses and characterizations of iron complexes of bulky o-phenylenediamide ligand
TMEDA in iron-catalyzed hydromagnesiation: Iron(II)-alkyl species for controlled reduction to alkene-stabilized iron(0)
The Exceptional Diversity of Homoleptic Uranium–Methyl Complexes
Peter G. N. Neate and Michael L. Neidig
Catalysis with Earth-abundant Elements, Uwe Schneider, Stephen Thomas, Eds., Royal Society of Chemistry, 2020, 231-245
Iron-catalysed reactions have seen extensive focus and development in recent years, due in part to increasing focus on sustainable methodologies. However, a significant challenge to this continued development is a lack of fundamental understanding of the active species and reaction pathways that govern reactivity in iron-catalysed systems. This chapter highlights the challenges in studying open shell iron catalysis as well as techniques that can be effectively used to achieve the desired molecular level insight. While these have provided substantial insight into what has long been regarded as a “black box”, both the strengths and limitations of these techniques are presented alongside highlights of potential pitfalls using recent literature examples.

Nikki J. Wolford, Xiaojuan Yu, Suzanne C. Bart, Jochen Autschbach and Michael L. Neidig
Dalton Trans. 2020, 49, 14401–14410
The trivalent oxidation state of uranium has been shown to undergo unique reactivity, from its ability to activate a variety of small molecules to its role in the catalytic reduction of ethene to ethane amongst others. Central to this unique reactivity and ability to rationally design ligands for isotope separation is the underlying uranium electronic structure. While electronic structure studies of U(IV), U(V), and U(VI) have been extensive, by comparison, analogous studies of more reduced oxidation states such as U(III) remains underdeveloped. Herein we report a combined MCD and EPR spectroscopic approach along with density functional theory and multireference wavefunction calculations to elucidate the effects of ligand perturbation in three uranium(III) Tp* complexes. Overall, the experimental and computational insight suggests that the change in ligand environment across this series of U(III) complexes resulted in only minor perturbations in the uranium electronic structure. This combined approach was also used to redefine the electronic ground state of a U(III) complex with a redox non-innocent Bipy− ligand. Overall, these studies demonstrate the efficacy of the combined experimental and theoretical approach towards evaluating electronic structure and bonding in U(III) complexes and provide important insight into the challenges in altering ligand environments to modify bonding and reactivity in uranium coordination chemistry.
Liu, R.; Ma, Z.; Sears, J. D.; Juneau, M.; Neidig, M. L.; Porosoff, M. D.
J. CO2 Util. 2020, 41, 101290
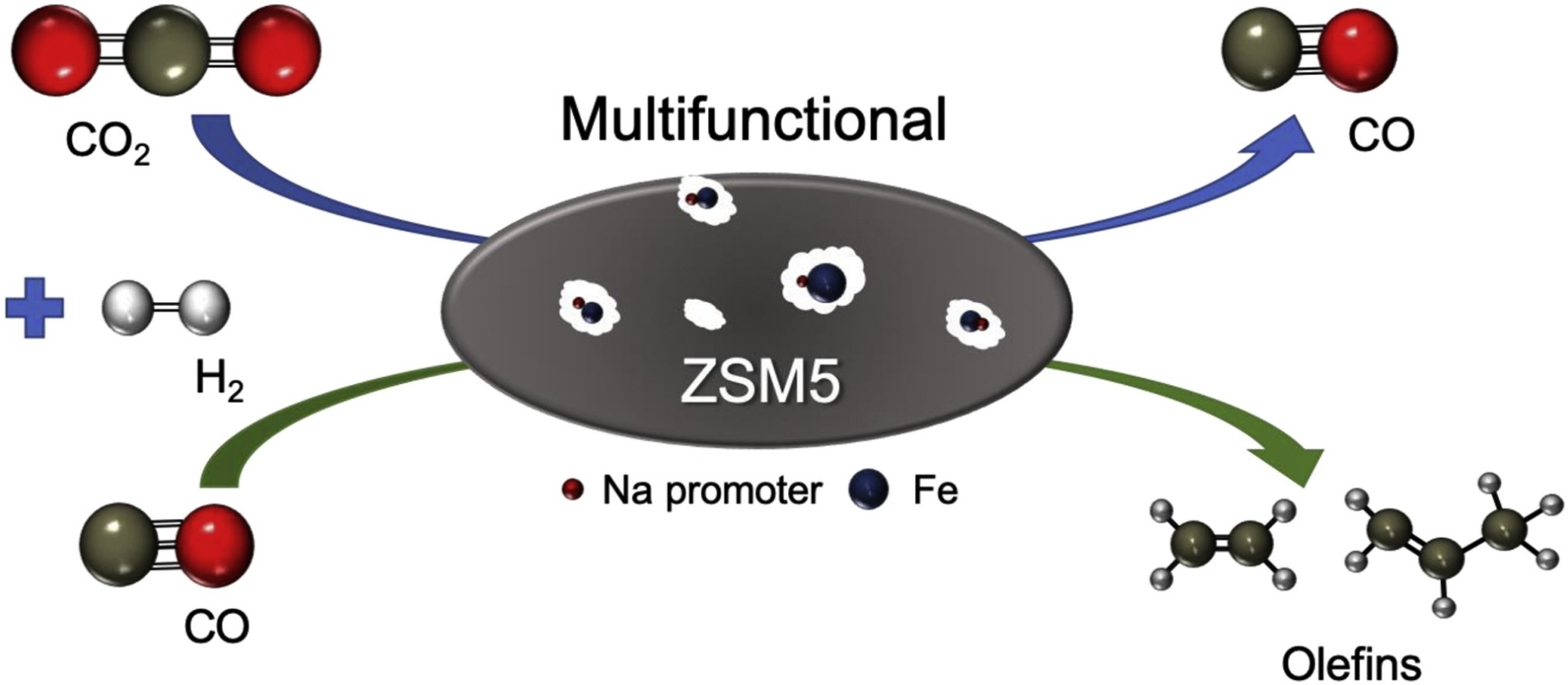
Correlations in Fischer-Tropsch synthesis (FTS) and CO2 hydrogenation are investigated over Fe supported on the acidic (H) and sodium (Na) forms of ZSM-5 (Si/Al = 50). FTS reactor studies indicate the selectivity toward olefins increases from 11% over Fe/H-ZSM-5 to 29.4% over Fe/Na-ZSM-5 because of Na increasing the surface basicity of the catalyst. Reactor studies are extended to CO2 hydrogenation, where reverse water-gas shift is the dominant reaction, with Fe/Na-ZSM-5 displaying enhanced CO2 adsorption, and in turn, higher CO selectivity (∼80%) versus Fe/H-ZSM-5 (∼60%). The catalysts are characterized by a variety of analytical techniques in-cluding Mössbauer spectroscopy, Fourier transform infrared (FTIR) spectroscopy and temperature programmed desorption (TPD) of NH3 and CO2 to correlate acid-base properties with catalytic performance. The findings of this study clearly show the selectivity of FTS and CO2 hydrogenation can be attenuated toward the desired products by modifying the acid-base properties of the catalyst with sodium. These results are an important step toward designing high-performance catalysts for light olefin synthesis from CO and CO2.
Liang, Q.; Lin, J. H.; DeMuth, J. C.; Neidig, M. L.; Song, D.
Dalton Trans. 2020, 49, 12287–12297
We report the syntheses of a family of tetrahedral iron complexes bearing a bulky redox active o-phenylenediamide ligand. The electronic structures of these complexes have been investigated by Mössbauer spectroscopy, magnetic susceptibility measurements, and X-ray crystallography.
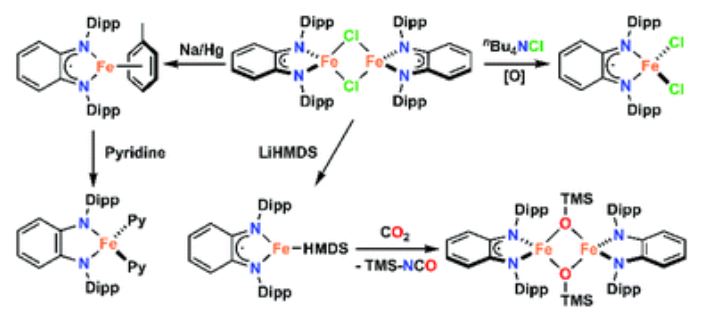
Neate, P. G. N.; Greenhalgh M. D.; Brennessel, W. W.; Thomas, S. P.; Neidig, M. L.
Angew. Chem. Int. Ed. 2020, 59, 17070–17076
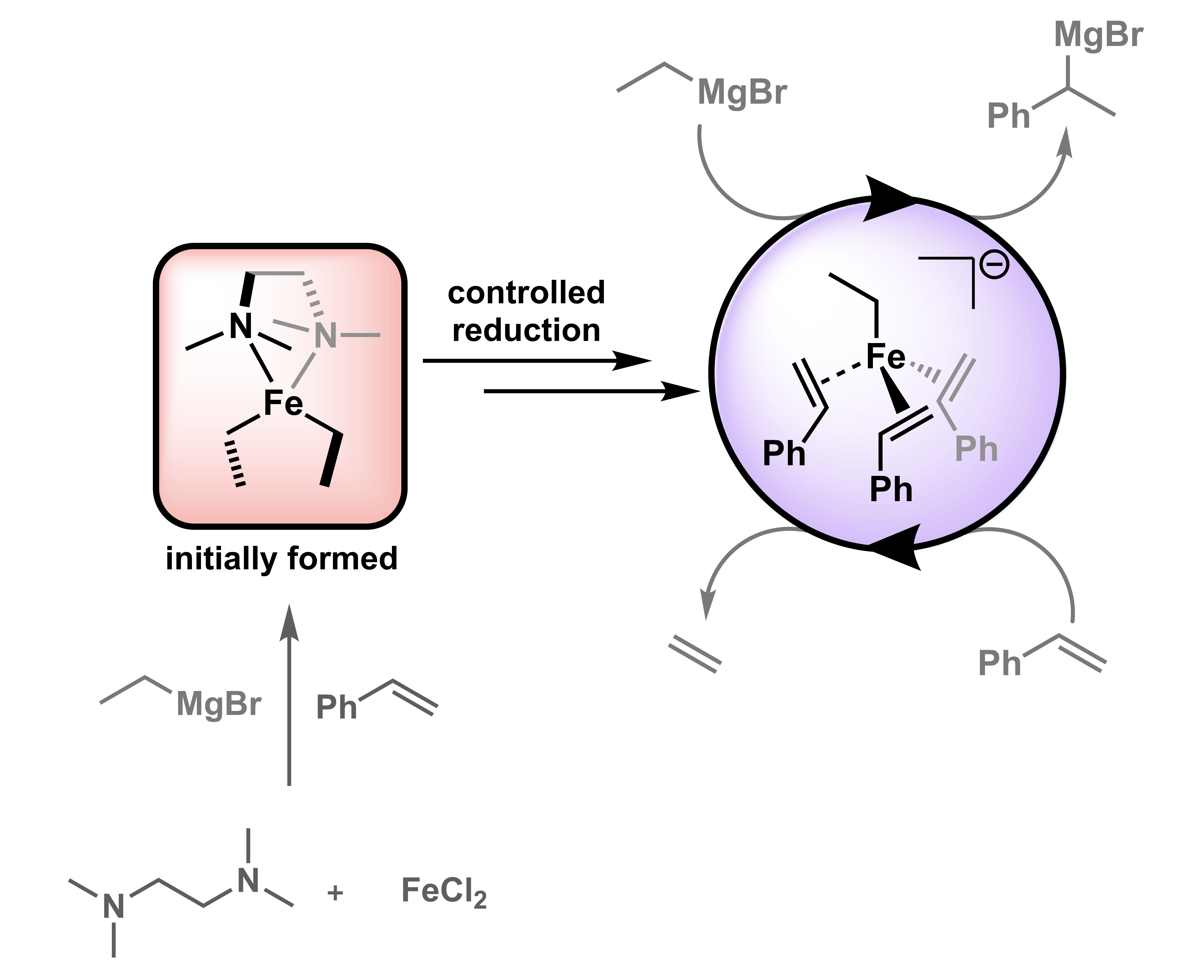
N,N,N′,N′‐Tetramethylethylenediamine (TMEDA) has been one of the most prevalent and successful additives used in iron catalysis, finding application in reactions as diverse as cross‐coupling, C−H activation, and borylation. However, the role that TMEDA plays in these reactions remains largely undefined. Herein, studying the iron‐catalyzed hydromagnesiation of styrene derivatives using TMEDA has provided molecular‐level insight into the role of TMEDA in achieving effective catalysis. The key is the initial formation of TMEDA–iron(II)–alkyl species which undergo a controlled reduction to selectively form catalytically active styrene‐stabilized iron(0)–alkyl complexes. While TMEDA is not bound to the catalytically active species, these active iron(0) complexes cannot be accessed in the absence of TMEDA. This mode of action, allowing for controlled reduction and access to iron(0) species, represents a new paradigm for the role of this important reaction additive in iron catalysis.
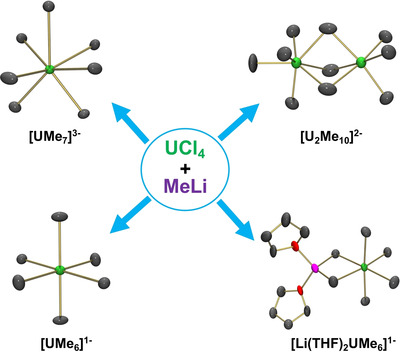
Jeffrey D. Sears, Dumitru‐Claudiu Sergentu, Tessa M. Baker, William W. Brennessel, Jochen Autschbach, Michael L. Neidig
Angew. Chem. Int. Ed. 2020, 59, 13586–13590
Homoleptic σ‐bonded uranium–alkyl complexes have been a synthetic target since the Manhattan Project. The current study describes the synthesis and characterization of several unprecedented uranium–methyl complexes. Amongst these complexes, the first example of a homoleptic uranium–alkyl dimer, [Li(THF)4]2[U2(CH3)10], as well as a seven‐coordinate uranium–methyl monomer, {Li(OEt2)Li(OEt2)2UMe7Li}n were both crystallographically identified. The diversity of complexes reported herein provides critical insight into the structural diversity, electronic structure and bonding in uranium–alkyl chemistry.
2019
Isolation and Characterization of a Homoleptic Tetramethylcobalt(III) Distorted Square-Planar Complex
Insight into the Electronic Structure of Formal Lanthanide(II) Complexes using Magnetic Circular Dichroism Spectroscopy
Atom-Economical Ni-Catalyzed Diborylative Cyclization of Enynes: Preparation of Unsymmetrical Diboronates
Identification and Reactivity of Cyclometalated Iron(II) Intermediates in Triazole-Directed Iron-Catalyzed C–H Activation
Mechanism of the Bis(imino)pyridine-Iron-Catalyzed Hydromagnesiation of Styrene Derivatives
Homoleptic Aryl Complexes of Uranium (IV)
Terminal coordination of diatomic boron monofluoride to iron
Crystal structure of bromidopentakis(tetrahydrofuran-κO)magnesium bis[1,2-bis(diphenylphosphanyl)benzene-κ2 P,P′]cobaltate(−1) tetrahydrofuran disolvate
The Effect of β‐Hydrogen Atoms on Iron Speciation in Cross‐Couplings with Simple Iron Salts and Alkyl Grignard Reagents
Development and Evolution of Mechanistic Understanding in Iron-Catalyzed Cross-Coupling
Reactive Intermediates and Mechanism in Iron‐Catalyzed Cross‐coupling
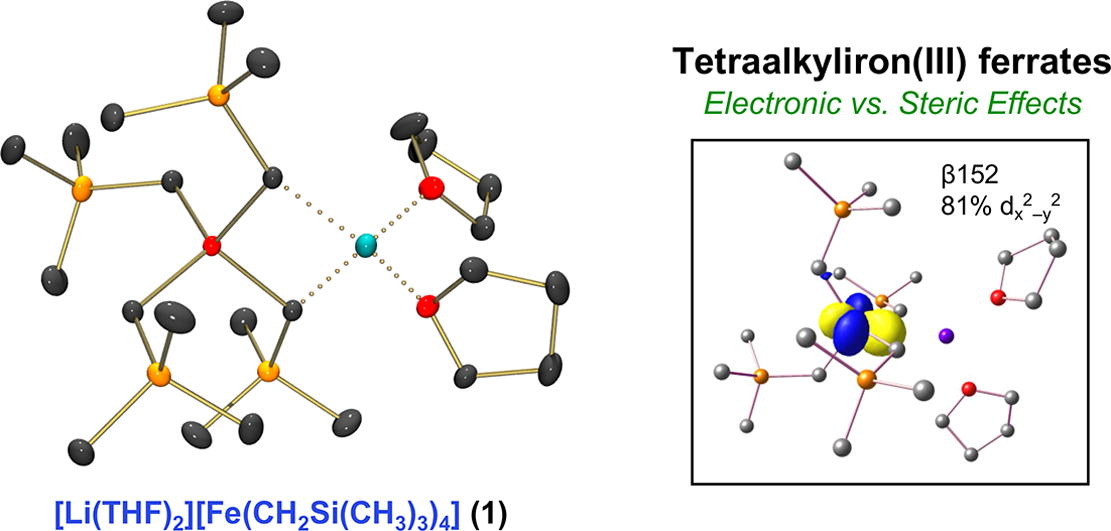
Synthesis and characterization of a sterically encumbered homoleptic tetraalkyliron(III) ferrate complex
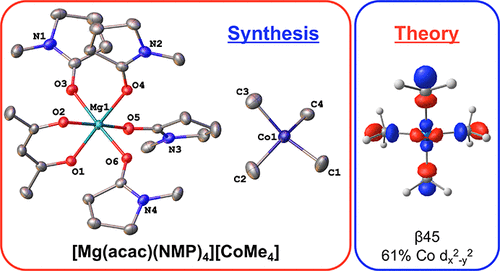
Stephanie H. Carpenter, William W. Brennessel, and Michael L. Neidig
Organometallics 2019, 38, 3486–3489
Homoleptic cobalt alkyl and aryl complexes are extremely rare, limited predominantly to complexes utilizing bulky, stabilizing ligands. There have been no reports to date of homoleptic cobalt species with simple, sterically unencumbered alkyl ligands, such as methyl. Herein, we report the synthesis and characterization of the homoleptic, distorted square-planar tetramethylcobalt(III) complex [Mg(acac)(NMP)4][CoMe4] (NMP = N-methylpyrrolidone), which exhibits extreme temperature and moisture sensitivities. NMR and density functional theory provide detailed insight into the electronic structure and bonding in this complex.
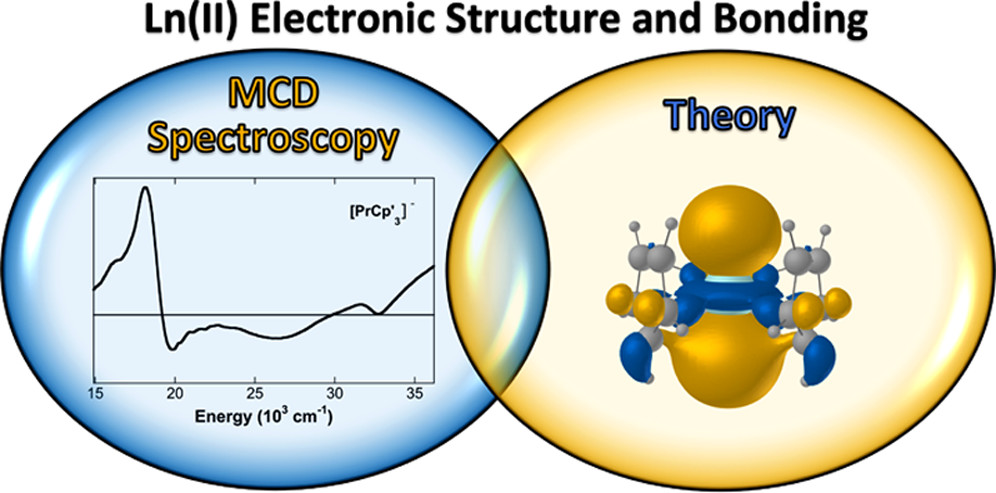
Valerie E. Fleischauer, Gaurab Ganguly, David H. Woen, Nikki J. Wolford, William J. Evans*, Jochen Autschbach* and Michael L. Neidig*
Organometallics 2019, 38, 3124-3131
Magnetic circular dichroism (MCD) spectroscopy has been utilized to evaluate the electronic structure of the tris(cyclopentadienyl) rare-earth complexes [K(2.2.2-cryptand)][LnCp′3] (Ln = Y, La, Pr, Eu, Gd; Cp′ = C5H4SiMe3), which contain ions in the formal +2 oxidation state. These complexes were chosen to evaluate the 4fn5d1 electron configuration assignments of the recently discovered La(II), Pr(II), and Gd(II) ions versus the traditional 4fn+1 configuration of the long-known Eu(II) ion. The 4d1 Y(II) complex provided another benchmark in the MCD study. Transitions with f-orbital character were observed in the NIR MCD spectra of the 4f25d1 complex [PrCp′3]−. This study provides the first direct observation of f–f transitions in such Ln(II) species. The broadening of these transition for Pr(II) provides further confirmation of the 4fn5d1 versus 4fn+1 electronic configurations previously proposed and supported by restricted active-space (RAS) calculations. For further insight into the electronic structure of these [LnCp′3]− complexes, experimental UV–vis MCD spectroscopy was coupled with spectral calculations, which allowed for the assignment of transitions. The sensitivity of UV–vis MCD to spin–orbit coupling (SOC) and the increased spectral resolution in comparison to electronic absorption spectroscopy enabled identification of low-energy nd to (n + 1)p transitions in this class of complexes. Combined, these studies provide further insight into the electronic transitions and overall electronic structure of low-valent lanthanide(II) organometallic complexes.
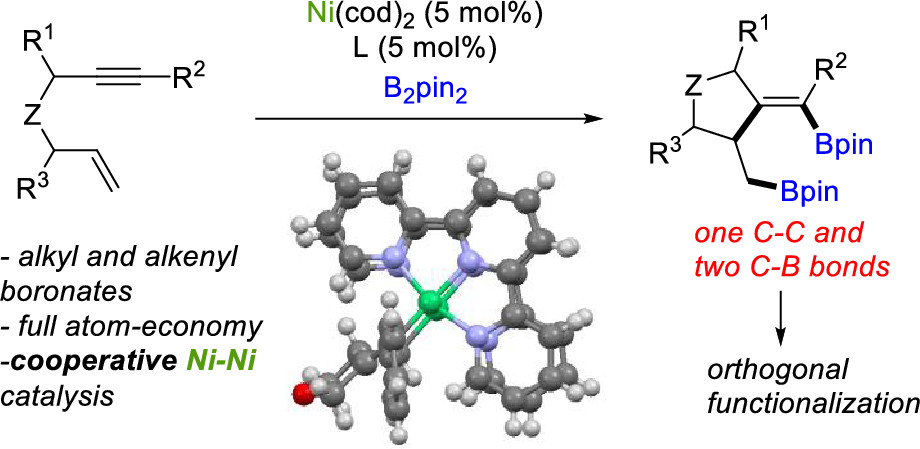
Natalia Cabrera-Lobera, M. Teresa Quirós, William W. Brennessel, Michael L. Neidig, Elena Buñuel* and Deigo J. Cárdenas*
Org. Lett. 2019, 21, 6552-6556
We report a Ni-catalyzed diborylative cyclization of enynes that affords carbo- and heterocycles containing both alkyl- and alkenylboronates. The reaction is fully atom-economical, shows a broad scope, and employs a powerful and inexpensive catalytic Ni-based system. The reaction mechanism seems to involve activation of the enyne by Ni(0) through oxidative cyclometalation of the enyne prior to diboron reagent activation. An unprecedented dinuclear bis(organometallic) Ni(I) intermediate complex was isolated.
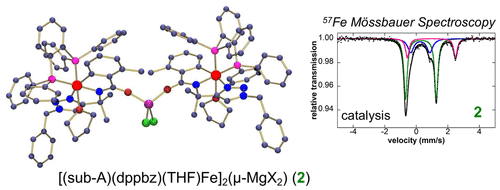
Theresa E. Boddie, Stephanie H. Carpenter, Tessa M. Baker, Joshua C. DeMuth, Gianpiero Cera, William W. Brennessel, Lutz Ackermann, and Michael L. Neidig*
J. Am. Chem. Soc. 2019, 141, 12338-12345
While iron-catalyzed C–H activation offers an attractive reaction methodology for organic transformations, the lack of molecular-level insight into the in situ formed and reactive iron species impedes continued reaction development. Herein, freeze-trapped 57Fe Mössbauer spectroscopy and single-crystal X-ray crystallography combined with reactivity studies are employed to define the key cyclometalated iron species active in triazole-assisted iron-catalyzed C–H activation. These studies provide the first direct experimental definition of an activated intermediate, which has been identified as the low-spin iron(II) complex [(sub-A)(dppbz)(THF)Fe]2(μ-MgX2), where sub-A is a deprotonated benzamide substrate. Reaction of this activated intermediate with additional diarylzinc leads to the formation of a cyclometalated iron(II)–aryl species, which upon reaction with oxidant, generates C–H arylated product at a catalytically relevant rate. Furthermore, pseudo-single-turnover reactions between catalytically relevant iron intermediates and excess nucleophile identify transmetalation as rate-determining, whereas C–H activation is shown to be facile under the reaction conditions.
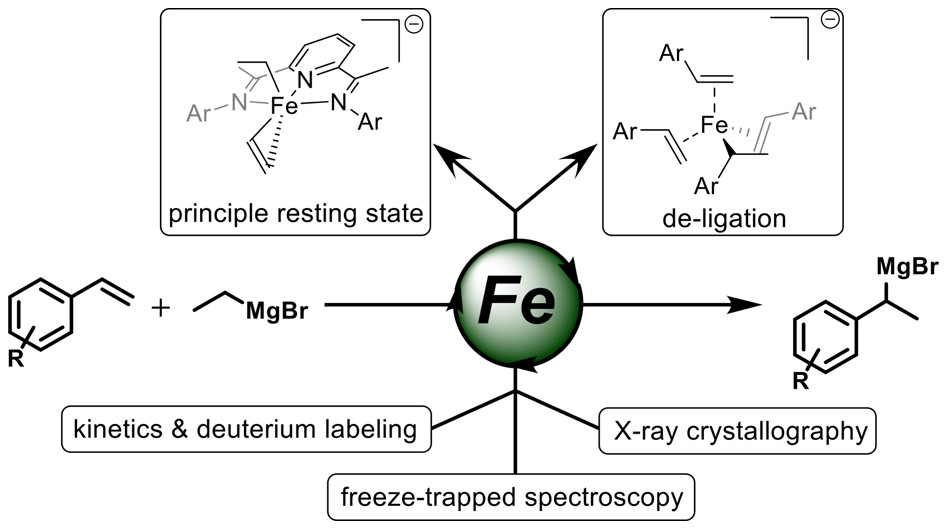
Peter G. N. Neate, Mark D. Greenhalgh, William W. Brennessel, Stephen P. Thomas*, and Michael L. Neidig*
J. Am. Chem. Soc. 2019, 141, 10099-10108
Iron-catalyzed hydromagnesiation of styrene derivatives offers a rapid and efficient method to generate benzylic Grignard reagents, which can be applied in a range of transformations to provide products of formal hydrofunctionalization. While iron-catalyzed methodologies exist for the hydromagnesiation of terminal alkenes, internal alkynes, and styrene derivatives, the underlying mechanisms of catalysis remain largely undefined. To address this issue and determine the divergent reactivity from established cross-coupling and hydrofunctionalization reactions, a detailed study of the bis(imino)pyridine iron-catalyzed hydromagnesiation of styrene derivatives is reported. Using a combination of kinetic analysis, deuterium labeling, and reactivity studies as well as in situ 57Fe Mössbauer spectroscopy, key mechanistic features and species were established. A formally iron(0) ate complex [iPrBIPFe(Et)(CH2═CH2)]− was identified as the principle resting state of the catalyst. Dissociation of ethene forms the catalytically active species which can reversibly coordinate the styrene derivative and mediate a direct and reversible β-hydride transfer, negating the necessity of a discrete iron hydride intermediate. Finally, displacement of the tridentate bis(imino)pyridine ligand over the course of the reaction results in the formation of a tris-styrene-coordinated iron(0) complex, which is also a competent catalyst for hydromagnesiation.
Nikki J. Wolford, Dumitru‐Claudiu Sergentu, William W. Brennessel, Jochen Autschbach*, and Michael L. Neidig*
Angew. Chem. Int. Ed. 2019, 58, 10266-10270
The synthesis and characterization of sterically unencumbered homoleptic organouranium aryl complexes containing U−C σ‐bonds has been of interest to the chemical community for over 70 years. Reported herein are the first structurally characterized, sterically unencumbered homoleptic uranium (IV) aryl‐ate species of the form [U(Ar)6]2− (Ar=Ph, p ‐tolyl, p ‐Cl‐Ph). Magnetic circular dichroism (MCD) spectroscopy and computational studies provide insight into electronic structure and bonding interactions in the U−C σ‐bond across this series of complexes. Overall, these studies solve a decades‐long challenge in synthetic uranium chemistry, enabling new insight into electronic structure and bonding in organouranium complexes.
Myles J. Drance, Jeffrey D. Sears, Anthony M. Mrse, Curtis E. Moore, Arnold L. Rheingold, Michael L. Neidig, Joshua S. Figueroa*
Science 2019, 363, 1203-1205
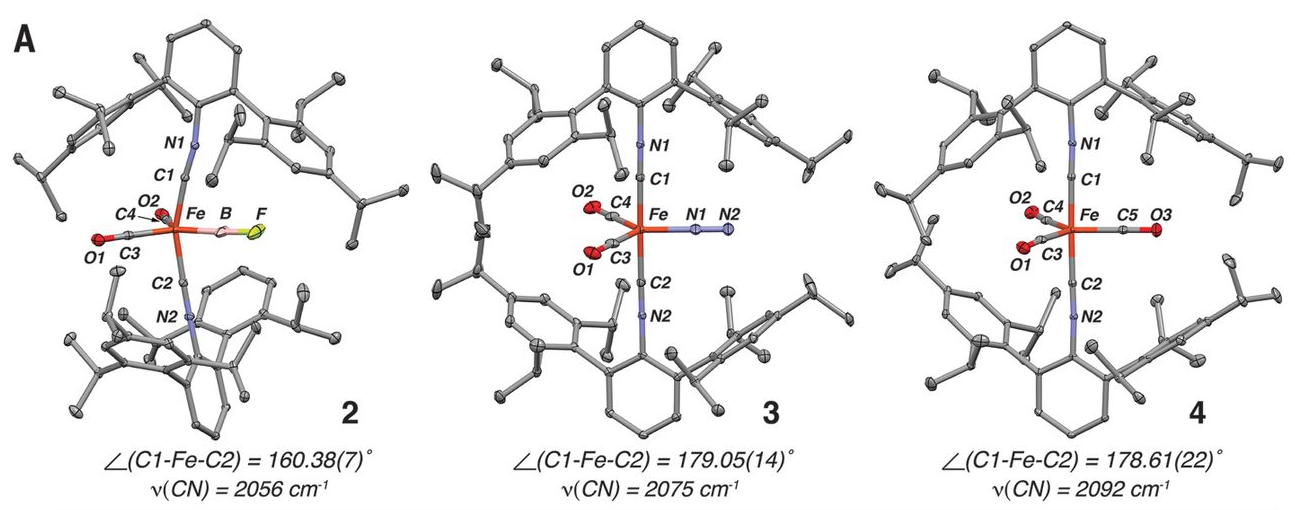
Boron monofluoride (BF) is a diatomic molecule with 10 valence electrons, isoelectronic to carbon monoxide (CO). Unlike CO, which is a stable molecule at room temperature and readily serves as both a bridging and terminal ligand to transition metals, BF is unstable below 1800°C in the gas phase, and its coordination chemistry is substantially limited. Here, we report the isolation of the iron complex Fe(BF)(CO)2(CNArTripp2)2 [ArTripp2, 2,6-(2,4,6-(i-Pr)3C6H2]2C6H3; i-Pr, iso-propyl], featuring a terminal BF ligand. Single-crystal x-ray diffraction as well as nuclear magnetic resonance, infrared, and Mössbauer spectroscopic studies on Fe(BF)(CO)2(CNArTripp2)2 and the isoelectronic dinitrogen (N2) and CO complexes Fe(N2)(CO)2(CNArTripp2)2 and Fe(CO)3(CNArTripp2)2 demonstrate that the terminal BF ligand possesses particularly strong σ-donor and π-acceptor properties. Density functional theory and electron-density topology calculations support this conclusion.
Patience B. Girigiri, Stephanie H. Carpenter, William W.Brennessel, And Michael L. Neidig*
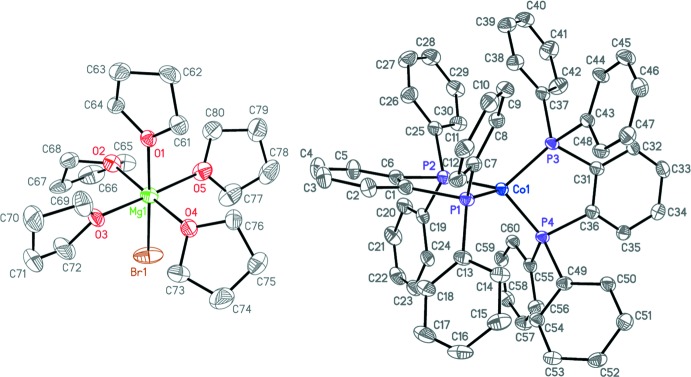
Acta Crystallogr E Crystallogr Commun. 2019, 75, 304-307
Structural characterization of the ionic title complex, [MgBr(THF)5][Co(dpbz)2]·2THF [THF is tetrahydrofuran, C4H8O; dpbz is 1,2-bis(diphenylphosphanyl)benzene, C30H24P2], revealed a well-separated cation and anion co-crystallized with two THF solvent molecules that interact with the cation via weak C—H⋯O contacts. The geometry about the cobalt center is pseudotetrahedral, as is expected for a d10 metal center, only deviating from an ideal tetrahedral geometry because of the restrictive bite angles of the bidentate phosphane ligands. Three THF ligands of the cation and one co-crystallized THF solvent molecule are each disordered over two orientations. In the extended structure, the cations and THF solvent molecules are arranged in (100) sheets that alternate with layers of anions, the latter of which show various π-interactions, which may explain the particular packing arrangement.
Jeffrey D. Sears, Dr. Salvador B. Muñoz III, Dr. Stephanie L. Daifuku, Ari A. Shaps, Stephanie H. Carpenter, Dr. William W. Brennessel, Prof. Michael L. Neidig*
Angew. Chem. Int. Ed. 2019, 58, 2769-2773
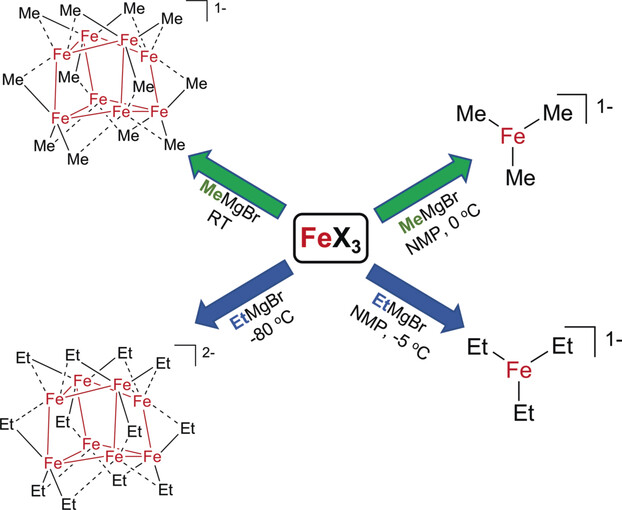
The effects of β‐hydrogen‐containing alkyl Grignard reagents in simple ferric salt cross‐couplings have been elucidated. The reaction of FeCl3 with EtMgBr in THF leads to the formation of the cluster species [Fe8Et12]2−, a rare example of a structurally characterized metal complex with bridging ethyl ligands. Analogous reactions in the presence of NMP, a key additive for effective cross‐coupling with simple ferric salts and β‐hydrogen‐containing alkyl nucleophiles, result in the formation of [FeEt3]−. Reactivity studies demonstrate the effectiveness of [FeEt3]− in rapidly and selectively forming the cross‐coupled product upon reaction with electrophiles. The identification of iron‐ate species with EtMgBr analogous to those previously observed with MeMgBr is a critical insight, indicating that analogous iron species can be operative in catalysis for these two classes of alkyl nucleophiles.
Michael L. Neidig*, Stephanie H. Carpenter, Daniel J. Curran, Joshua C. DeMuth, Valerie E. Fleischauer, Theresa E. Iannuzzi, Peter G. N. Neate, Jeffrey D. Sears, and Nikki J. Wolford
Acc. Chem. Res. 2019, 52, 140-150
Since the pioneering work of Kochi in the 1970s, iron has attracted great interest for cross-coupling catalysis due to its low cost and toxicity as well as its potential for novel reactivity compared to analogous reactions with precious metals like palladium. Today there are numerous iron-based cross-coupling methodologies available, including challenging alkyl–alkyl and enantioselective methods. Furthermore, cross-couplings with simple ferric salts and additives like NMP and TMEDA (N-methylpyrrolidone and tetramethylethylenediamine) continue to attract interest in pharmaceutical applications. Despite the tremendous advances in iron cross-coupling methodologies, in situ formed and reactive iron species and the underlying mechanisms of catalysis remain poorly understood in many cases, inhibiting mechanism-driven methodology development in this field. This lack of mechanism-driven development has been due, in part, to the challenges of applying traditional characterization methods such as nuclear magnetic resonance (NMR) spectroscopy to iron chemistry due to the multitude of paramagnetic species that can form in situ. The application of a broad array of inorganic spectroscopic methods (e.g., electron paramagnetic resonance, 57Fe Mössbauer, and magnetic circular dichroism) removes this barrier and has revolutionized our ability to evaluate iron speciation. In conjunction with inorganic syntheses of unstable organoiron intermediates and combined inorganic spectroscopy/gas chromatography studies to evaluate in situ iron reactivity, this approach has dramatically evolved our understanding of in situ iron speciation, reactivity, and mechanisms in iron-catalyzed cross-coupling over the past 5 years. This Account focuses on the key advances made in obtaining mechanistic insight in iron-catalyzed carbon–carbon cross-couplings using simple ferric salts, iron-bisphosphines, and iron-N-heterocyclic carbenes (NHCs). Our studies of ferric salt catalysis have resulted in the isolation of an unprecedented iron-methyl cluster, allowing us to identify a novel reaction pathway and solve a decades-old mystery in iron chemistry. NMP has also been identified as a key to accessing more stable intermediates in reactions containing nucleophiles with and without β-hydrogens. In iron-bisphosphine chemistry, we have identified several series of transmetalated iron(II)-bisphosphine complexes containing mesityl, phenyl, and alkynyl nucleophile-derived ligands, where mesityl systems were found to be unreliable analogues to phenyls. Finally, in iron-NHC cross-coupling, unique chelation effects were observed in cases where nucleophile-derived ligands contained coordinating functional groups. As with the bisphosphine case, high-spin iron(II) complexes were shown to be reactive and selective in cross-coupling. Overall, these studies have demonstrated key aspects of iron cross-coupling and the utility of detailed speciation and mechanistic studies for the rational improvement and development of iron cross-coupling methods.
Kneebone, J. K.; Sears, J. D.; Neidig, M. L.
Non-Noble Metal Catalysis, R.J.M. Klein Gebbink, M.-E. Moret, Eds., Wiley-VCH: Weinheim, 2019, 265-295
The effects of β‐hydrogen‐containing alkyl Grignard reagents in simple ferric salt cross‐couplings have been elucidated. The reaction of FeCl3 with EtMgBr in THF leads to the formation of the cluster species [Fe8Et12]2−, a rare example of a structurally characterized metal complex with bridging ethyl ligands. Analogous reactions in the presence of NMP, a key additive for effective cross‐coupling with simple ferric salts and β‐hydrogen‐containing alkyl nucleophiles, result in the formation of [FeEt3]−. Reactivity studies demonstrate the effectiveness of [FeEt3]− in rapidly and selectively forming the cross‐coupled product upon reaction with electrophiles. The identification of iron‐ate species with EtMgBr analogous to those previously observed with MeMgBr is a critical insight, indicating that analogous iron species can be operative in catalysis for these two classes of alkyl nucleophiles.

Jeffrey D. Sears, Salvador B. Muñoz III, Maria Camila AguileraCuenca, William W. Brennessel, Michael L. Neidig*
Polyhedron 2019, 158, 91-96
The effects of β‐hydrogen‐containing alkyl Grignard reagents in simple ferric salt cross‐couplings have been elucidated. The reaction of FeCl3 with EtMgBr in THF leads to the formation of the cluster species [Fe8Et12]2−, a rare example of a structurally characterized metal complex with bridging ethyl ligands. Analogous reactions in the presence of NMP, a key additive for effective cross‐coupling with simple ferric salts and β‐hydrogen‐containing alkyl nucleophiles, result in the formation of [FeEt3]−. Reactivity studies demonstrate the effectiveness of [FeEt3]− in rapidly and selectively forming the cross‐coupled product upon reaction with electrophiles. The identification of iron‐ate species with EtMgBr analogous to those previously observed with MeMgBr is a critical insight, indicating that analogous iron species can be operative in catalysis for these two classes of alkyl nucleophiles.
2018
Intermediates and Mechanism in Iron-Catalyzed Cross-Coupling
Combined Effects of Backbone and N-Substituents on Structure, Bonding, and Reactivity of Alkylated Iron(II)-NHCs
Multinuclear iron–phenyl species in reactions of simple iron salts with PhMgBr: identification of Fe4(μ-Ph)6(THF)4 as a key reactive species for cross-coupling catalysis
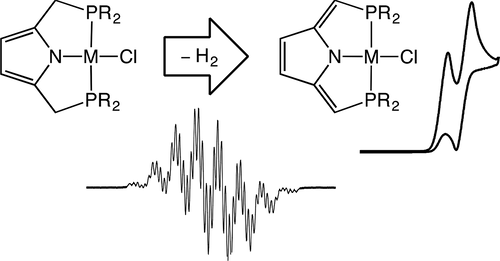
Backbone Dehydrogenation in Pyrrole-Based Pincer Ligands
Nitric oxide activation facilitated by cooperative multimetallic electron transfer within an iron-functionalized polyoxovanadate–alkoxide cluster
A Pseudotetrahedral Uranium(V) Complex
Crystal structures of two new six-coordinate iron(III) complexes with 1,2-bis(diphenylphosphane) ligands
The N ‐Methylpyrrolidone (NMP) Effect in Iron‐Catalyzed Cross‐Coupling with Simple Ferric Salts and MeMgBr
NHC and nucleophile chelation effects on reactive iron(II) species in alkyl–alkyl cross-coupling
Jeffrey D. Sears, Peter G. N. Neate, and Michael L. Neidig*
J. Am. Chem. Soc. 2018, 140, 11872-11883
Iron-catalyzed cross-coupling reactions have attracted significant research interest, as they offer numerous favorable features compared with cross-coupling reactions with precious metal catalysis. While this research has contributed to an empirical understanding of iron-catalyzed cross-coupling, the underlying fundamental mechanisms of reaction and structures of catalytically active species have remained poorly defined. The lack of such detail can be attributed to the difficulties associated with studying such iron-catalyzed reactions, where unstable paramagnetic intermediates abound. Recently, the combined application of physical-inorganic spectroscopic methods, concomitant organic product analysis, and air- and temperature-sensitive inorganic synthesis has yielded the most detailed insight currently available on reactivity and mechanism in iron-catalyzed cross-coupling. This Perspective highlights this approach and the limitations of the contributing techniques as well as some of the key features of the catalytic reactions studied and lessons learned.
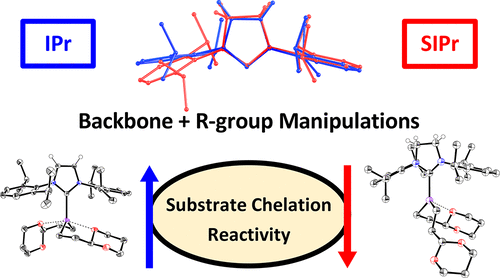
Salvador B. Muñoz, III, Valerie E. Fleischauer, William W. Brennessel, and Michael L. Neidig*
Organometallics 2018, 37, 3093-3101
Iron and N-heterocyclic carbenes (NHCs) have proven to be a successful pair in catalysis, with reactivity and selectivity being highly dependent on the nature of the NHC ligand backbone saturation and N-substituents. Four (NHC)Fe(1,3-dioxan-2-ylethyl)2 complexes have been isolated and spectroscopically characterized to correlate their reactivity to steric effects of the NHC from both the backbone saturation and N-substituents. Only in the extreme case of SIPr where NHC backbone and N-substituent steric effects are the largest is there a major structural perturbation observed crystallographically. The addition of only two hydrogen atoms is sufficient for a drastic change in product selectivity in the coupling of 1-iodo-3-phenylpropane with (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide due to resulting structural perturbations to the precatalyst. Mössbauer spectroscopy and magnetic circular dichroism enabled the correlation of covalency and steric bulk in the SIPr case to its poor selectivity in alkyl–alkyl cross-coupling with iron. Density functional theory calculations provided insight into the electronic structure and molecular orbital effects of ligation changes to the iron center. Finally, charge donation analysis and Mayer bond order calculations further confirmed the stronger Fe–ligand bonding in the SIPr complex. Overall, these studies highlight the importance of considering both N-substituent and backbone steric contributions to structure, bonding, and reactivity in iron-NHCs.
Stephanie H. Carpenter, Tessa M. Baker, Salvador B. Muñoz III, William W. Brennessel and Michael L. Neidig*
Chem. Sci. 2018, 9, 7931-7939
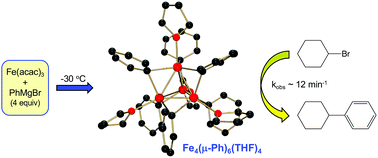
The first direct syntheses, structural characterizations, and reactivity studies of iron–phenyl species formed upon reaction of Fe(acac)3 and PhMgBr in THF are presented. Reaction of Fe(acac)3 with 4 equiv. PhMgBr in THF leads to the formation of [FePh2(μ-Ph)]22− at −80 °C, which can be stabilized through the addition of N-methylpyrrolidone. Alternatively, at −30 °C this reaction leads to the formation of the tetranuclear iron–phenyl cluster, Fe4(μ-Ph)6(THF)4. Further synthetic studies demonstrate that analogous tetranuclear iron clusters can be formed with both 4-F-PhMgBr and p-tolylMgBr, illustrating the generality of this structural motif for reactions of simple ferric salts and aryl Grignard reagents in THF. Additional studies isolate and define key iron species involved in the synthetic pathway leading to the formation of the tetranuclear iron–aryl species. While reaction studies demonstrate that [FePh2(μ-Ph)]22− is unreactive towards electrophile, Fe4(μ-Ph)6(THF)4 is found to rapidly react with bromocyclohexane to selectively form phenylcyclohexane. Based on this reactivity, a new catalytic reaction protocol has been developed that enables efficient cross-couplings using Fe4(μ-Ph)6(THF)4, circumventing the current need for additives such as TMEDA or supporting ligands to achieve effective cross-coupling of PhMgBr and a secondary alkyl halide.
V. Mahesh Krishnan, Ian Davis, Tessa M. Baker, Daniel J. Curran, Hadi D. Arman, Michael L. Neidig, Aimin Liu, and Zachary J. Tonzetich
Inorg. Chem. 2018, 57, 9544-9553
The first direct syntheses, structural characterizations, and reactivity studies of iron–phenyl species formed upon reaction of Fe(acac)3 and PhMgBr in THF are presented. Reaction of Fe(acac)3 with 4 equiv. PhMgBr in THF leads to the formation of [FePh2(μ-Ph)]22− at −80 °C, which can be stabilized through the addition of N-methylpyrrolidone. Alternatively, at −30 °C this reaction leads to the formation of the tetranuclear iron–phenyl cluster, Fe4(μ-Ph)6(THF)4. Further synthetic studies demonstrate that analogous tetranuclear iron clusters can be formed with both 4-F-PhMgBr and p-tolylMgBr, illustrating the generality of this structural motif for reactions of simple ferric salts and aryl Grignard reagents in THF. Additional studies isolate and define key iron species involved in the synthetic pathway leading to the formation of the tetranuclear iron–aryl species. While reaction studies demonstrate that [FePh2(μ-Ph)]22− is unreactive towards electrophile, Fe4(μ-Ph)6(THF)4 is found to rapidly react with bromocyclohexane to selectively form phenylcyclohexane. Based on this reactivity, a new catalytic reaction protocol has been developed that enables efficient cross-couplings using Fe4(μ-Ph)6(THF)4, circumventing the current need for additives such as TMEDA or supporting ligands to achieve effective cross-coupling of PhMgBr and a secondary alkyl halide.
Feng Li, Rachel L. Meyer, Stephanie H. Carpenter, Lauren E. VanGelder, Asa. W. Nichols, Charles. W. Machan, Michael L. Neidig, and Ellen M. Matson
Chem. Sci. 2018, 9, 6379-6389
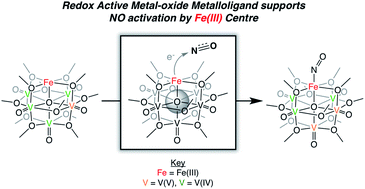
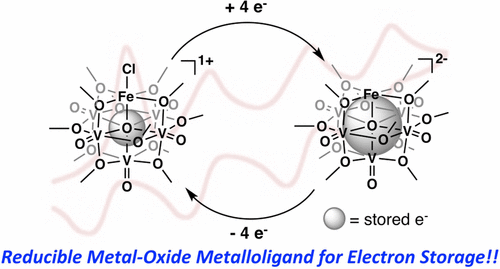
A series of NO-bound, iron-functionalized polyoxovanadate–alkoxide (FePOV–alkoxide) clusters have been synthesized, providing insight into the role of multimetallic constructs in the coordination and activation of a substrate. Upon exposure of the heterometallic cluster to NO, the vanadium-oxide metalloligand is oxidized by a single electron, shuttling the reducing equivalent to the {FeNO} subunit to form a {FeNO}7 species. Four NO-bound clusters with electronic distributions ranging from [VV3VIV2]{FeNO}7 to [VIV5]{FeNO}7 have been synthesized, and characterized via1H NMR, infrared, and electronic absorption spectroscopies. The ability of the FePOV–alkoxide cluster to store reducing equivalents in the metalloligand for substrate coordination and activation highlights the ultility of the metal-oxide scaffold as a redox reservoir.
Aaron M. Tondreau, Thomas J. Duignan, Benjamin W. Stein, Valerie E. Fleischauer, Jochen Autschbach, Enrique R. Batista, James M. Boncella, Maryline G. Ferrier, Stosh A. Kozimor, Veronika Mocko, Michael L. Neidig, Samantha K. Cary, and Ping Yang
Inorg. Chem. 2018, 57, 8106-8115
A series of uranium amides were synthesized from N,N,N-cyclohexyl(trimethylsilyl)lithium amide [Li][N(TMS)Cy] and uranium tetrachloride to give U(NCySiMe3)x(Cl)4–x, where x = 2, 3, or 4. The diamide was isolated as a bimetallic, bridging lithium chloride adduct ((UCl2(NCyTMS)2)2-LiCl(THF)2), and the tris(amide) was isolated as the lithium chloride adduct of the monometallic species (UCl(NCyTMS)3-LiCl(THF)2). The tetraamide complex was isolated as the four-coordinate pseudotetrahedron. Cyclic voltammetry revealed an easily accessible reversible oxidation wave, and upon chemical oxidation, the UV amido cation was isolated in near-quantitative yields. The synthesis of this family of compounds allows a direct comparison of the electronic structure and properties of isostructural UIV and UV tetraamide complexes. Spectroscopic investigations consisting of UV–vis, NIR, MCD, EPR, and U L3-edge XANES, along with density functional and wave function calculations, of the four-coordinate UIV and UVcomplexes have been used to understand the electronic structure of these pseudotetrahedral complexes.
Derek L. McNeil Jr., Daihlia J. Beckford, Jared L. Kneebone, Stephanie H. Carpenter, William W. Brennessel and Michael L. Neidig
Acta Crystallogr E Crystallogr Commun. 2018, 74, 803-807
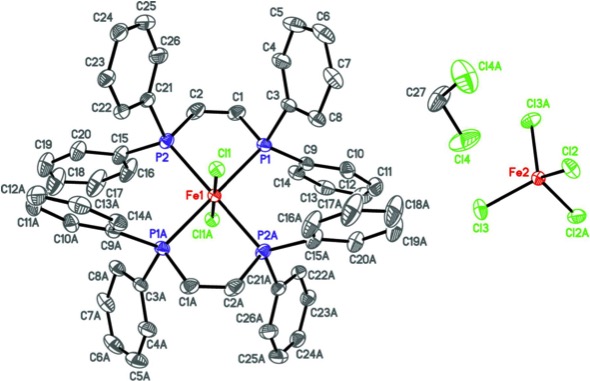
Structural characterization of the ionic complexes [FeCl2(C26H22P2)2][FeCl4]·0.59CH2Cl2 or [(dppen)2FeCl2][FeCl4]·0.59CH2Cl2 (dppen = cis-1,2-bis(diphenylphosphane)ethylene, P2C26H22) and [FeCl2(C30H24P2)2][FeCl4]·CH2Cl2 or [(dpbz)2FeCl2][FeCl4]·CH2Cl2 (dpbz = 1,2-bis(diphenylphosphane)benzene, P2C30H24) demonstrates trans coordination of two bidentate phosphane ligands (bisphosphanes) to a single iron(III) center, resulting in six-coordinate cationic complexes that are balanced in charge by tetrachloridoferrate(III) monoanions. The trans bisphosphane coordination is consistent will all previously reported molecular structures of six coordinate iron(III) complex cations with a (PP)2X2 (X = halido) donor set. The complex with dppen crystallizes in the centrosymmetric space group C2/c as a partial-occupancy [0.592 (4)] dichloromethane solvate, while the dpbz-ligated complex crystallizes in the triclinic space group P1 as a full dichloromethane monosolvate. Furthermore, the crystal studied of [(dpbz)2FeCl2][FeCl4]·CH2Cl2 was an inversion twin, whose component mass ratio refined to 0.76 (3):0.24 (3). Beyond a few very weak C—H⋯Cl and C—H⋯π interactions, there are no significant supramolecular features in either structure.
Salvador B. Muñoz III, Stephanie L. Daifuku, Jeffrey D. Sears, Tessa M. Baker, Stephanie H. Carpenter, William W. Brennessel and Michael L. Neidig
Angew. Chem. Int. Ed. 2018, 57, 6496-6500
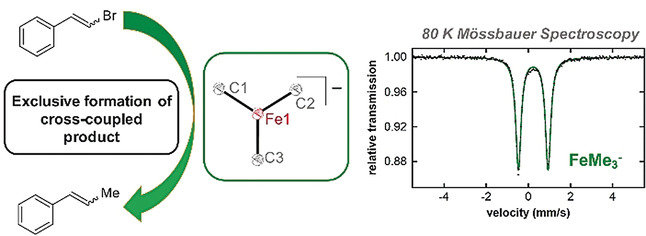
The use of N ‐methylpyrrolidone (NMP) as a co‐solvent in ferric salt catalyzed cross‐coupling reactions is crucial for achieving the highly selective, preparative scale formation of cross‐coupled product in reactions utilizing alkyl Grignard reagents. Despite the critical importance of NMP, the molecular level effect of NMP on in situ formed and reactive iron species that enables effective catalysis remains undefined. Herein, we report the isolation and characterization of a novel trimethyliron(II) ferrate species, [Mg(NMP)6][FeMe3]2 (1), which forms as the major iron species in situ in reactions of Fe(acac)3 and MeMgBr under catalytically relevant conditions where NMP is employed as a co‐solvent. Importantly, combined GC analysis and 57Fe Mössbauer spectroscopic studies identified 1 as a highly reactive iron species for the selective formation generating cross‐coupled product. These studies demonstrate that NMP does not directly interact with iron as a ligand in catalysis but, alternatively, interacts with the magnesium cations to preferentially stabilize the formation of 1 over [Fe8Me12]− cluster generation, which occurs in the absence of NMP.
Valerie E. Fleischauer, Salvador B. Muñoz III, Peter G. N. Neate, William W. Brennessel, and Michael L. Neidig
Chem. Sci. 2018, 9, 1878-1891
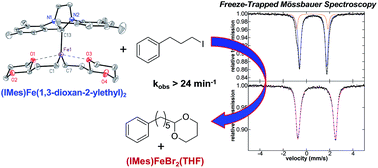
While iron–NHC catalysed cross-couplings have been shown to be effective for a wide variety of reactions (e.g. aryl–aryl, aryl–alkyl, alkyl–alkyl), the nature of the in situ formed and reactive iron species in effective catalytic systems remains largely undefined. In the current study, freeze-trapped Mössbauer spectroscopy, and EPR studies combined with inorganic synthesis and reaction studies are utilised to define the key in situ formed and reactive iron–NHC species in the Kumada alkyl–alkyl cross-coupling of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide and 1-iodo-3-phenylpropane. The key reactive iron species formed in situ is identified as (IMes)Fe((1,3-dioxan-2-yl)ethyl)2, whereas the S = 1/2 iron species previously identified in this chemistry is found to be only a very minor off-cycle species (<0.5% of all iron). Reaction and kinetic studies demonstrate that (IMes)Fe((1,3-dioxan-2-yl)ethyl)2 is highly reactive towards the electrophile resulting in two turnovers with respect to iron (kobs > 24 min−1) to generate cross-coupled product with overall selectivity analogous to catalysis. The high resistance of this catalytic system to β-hydride elimination of the alkyl nucleophile is attributed to its chelation to iron through ligation of carbon and one oxygen of the acetal moiety of the nucleophile. In fact, alternative NHC ligands such as SIPr are less effective in catalysis due to their increased steric bulk inhibiting the ability of the alkyl ligands to chelate. Overall, this study identifies a novel alkyl chelation method to achieve effective alkyl–alkyl cross-coupling with iron(II)–NHCs, provides direct structural insight into NHC effects on catalytic performance and extends the importance of iron(II) reactive species in iron-catalysed cross-coupling.
2017
Iron(II) Complexes of a Hemilabile SNS Amido Ligand: Synthesis, Characterization, and Reactivity
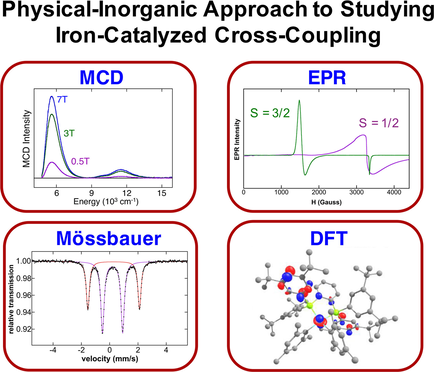
A Physical‐Inorganic Approach for the Elucidation of Active Iron Species and Mechanism in Iron‐Catalyzed Cross‐Coupling
Magnetic circular dichroism and density functional theory studies of electronic structure and bonding in cobalt(II)–N-heterocyclic carbene complexes
A Combined Probe-Molecule, Mössbauer, Nuclear Resonance Vibrational Spectroscopy, and Density Functional Theory Approach for Evaluation of Potential Iron Active Sites in an Oxygen Reduction Reaction Catalyst
Polyoxovanadate–Alkoxide Clusters as a Redox Reservoir for Iron
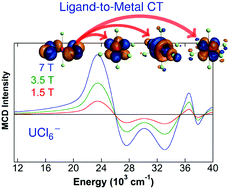
Magnetic circular dichroism of UCl6− in the ligand-to-metal charge-transfer spectral region
Intermediates and Reactivity in Iron-Catalyzed Cross-Couplings of Alkynyl Grignards with Alkyl Halides
Transition-Metal-Free Formation of C–E Bonds (E = C, N, O, S) and Formation of C–M Bonds (M = Mn, Mo) from N-Heterocyclic Carbene Mediated Fluoroalkene C–F Bond Activation
Magnetic circular dichroism studies of iron(ii) binding to human calprotectin
Uttam K. Das, Stephanie L. Daifuku, Theresa E. Iannuzzi, Serge I. Gorelsky, Ilia Korobkov, Bulat Gabidullin, Michael L. Neidig, and R. Tom Baker
Inorg. Chem. 2017, 56, 13766-13776
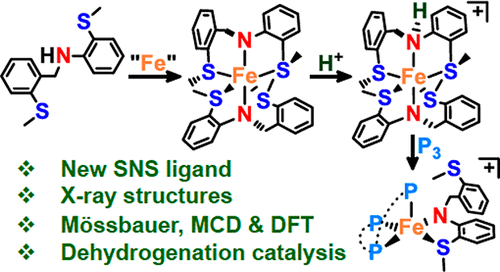
We report an easily prepared bis(thioether) amine ligand, SMeNHSMe, along with the synthesis, characterization, and reactivity of the paramagnetic iron(II) bis(amido) complex, [Fe(κ3-SMeNHSMe)2] (1). Binding of the two different thioethers to Fe generates both five- and six-membered rings with Fe–S bonds in the five-membered rings (av 2.54 Å) being significantly shorter than those in the six-membered rings (av 2.71 Å), suggesting hemilability of the latter thioethers. Consistent with this hypothesis, magnetic circular dichroism (MCD) and computational (TD-DFT) studies indicate that 1 in solution contains a five-coordinate component [Fe(κ3-SMeNHSMe)(κ2-SMeNHSMe)] (2). This ligand hemilability was demonstrated further by reactivity studies of 1 with 2,2′-bipyridine, 1,2-bis(dimethylphosphino)ethane, and 2,6-dimethylphenyl isonitrile to afford iron(II) complexes [L2Fe(κ2-SMeNHSMe)2] (3–5). Addition of a Brønsted acid, HNTf2, to 1 produces the paramagnetic, iron(II) amine–amido cation, [Fe(κ3-SMeNHSMe)(κ3-SMeNHSMe)](NTf2) (6; Tf = SO2CF3). Cation 6 readily undergoes amine ligand substitution by triphos, affording the 16e– complex [Fe(κ2-SMeNHSMe)(κ3-triphos)](NTf2) (7; triphos = bis(2-diphenylphosphinoethyl)phenylphosphine). These complexes are characterized by elemental analysis; 1H NMR, Mössbauer, IR, and UV–vis spectroscopy; and single-crystal X-ray diffraction. Preliminary results of amine–borane dehydrogenation catalysis show complex 7 to be a selective and particularly robust precatalyst.
Stephanie H. Carpenter, and Michael L. Neidig
Isr. J. Chem. 2017, 57, 1106-1116
Detailed studies of iron speciation and mechanism in iron‐catalyzed cross‐coupling reactions are critical for providing the necessary fundamental insight to drive new reaction development. However, such insight is challenging to obtain due to the prevalence of mixtures of unstable, paramagnetic organoiron species that can form in this chemistry. A physical‐inorganic research approach combining freeze‐trapped inorganic spectroscopic studies, organometallic synthesis and GC/kinetic studies provides a powerful method for studying such systems. Mössbauer, EPR and MCD spectroscopy enable the direct investigation of in situ formed iron species and, combined with GC analysis, the direct correlation of reactions of specific iron species to the generation of organic products. This review focuses on a description of the key methods involved in this physical‐inorganic approach, as well as examples of its application to investigations of iron‐SciOPP catalyzed cross‐coupling catalysis.
Theresa E. Iannuzzi, Yafei Gao, Tessa M. Baker, Liang Deng, and Michael L. Neidig
Dalton Trans. 2017, 46, 13290-13299
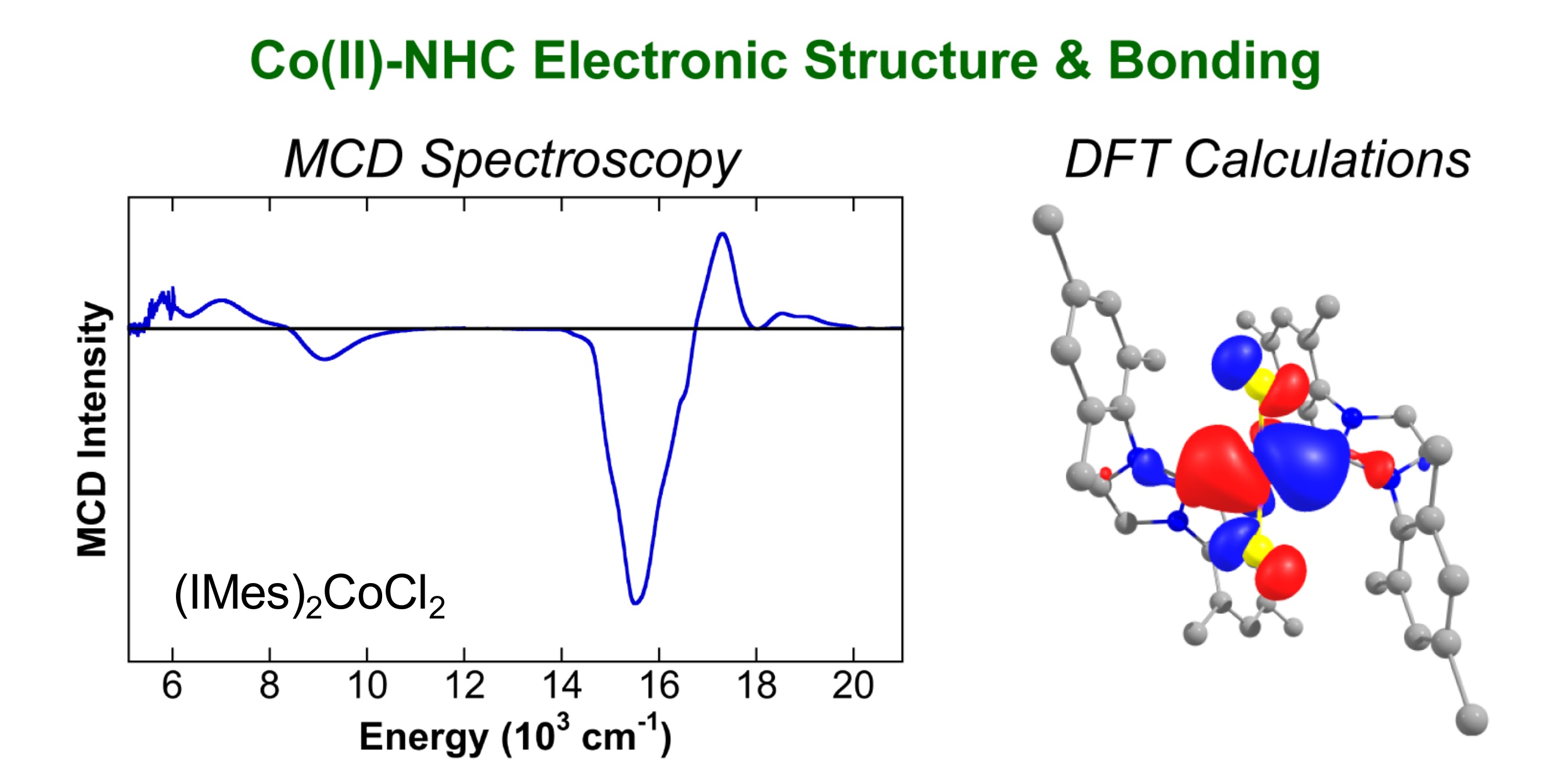
The combination of simple cobalt salts and N-heterocyclic carbene (NHC) ligands has been highly effective in C–H functionalization, hydroarylation and cross-coupling catalysis, though displaying a strong dependence on the identity of the NHC ligand. In addition, reactions effective with NHC ligands are often ineffective with phosphine ligands, further motivating the evaluation of the fundamental electronic structure and bonding differences in well-defined distorted tetrahedral Co(II) complexes. Magnetic circular dichroism (MCD) studies indicate that Co(II)–bisphosphines have larger ligand fields than Co(II)–NHC complexes. Theoretical density functional theory (DFT) calculations were performed on an expanded set of L2CoCl2 complexes (L2 = NHC, bisphosphine and diamine) to study the electronic structure and relative ligation properties of NHCs compared to bisphosphine and diamine ligands. Mayer bond order and charge decomposition analyses indicate that NHC ligands are slightly stronger donor ligands than bisphosphines but also result in a weakening of Co–Cl bonds in a trans-like influence. From MCD and DFT studies, changing the NHC N-substituent has a larger effect on the ligand field of Co(II)–NHC complexes than saturating the backbone. Overall, these studies provide detailed insight into the electronic structure and bonding effects in Co(II) complexes with ligand types commonly explored in catalysis.
Jared L. Kneebone, Stephanie L. Daifuku, Jeffrey A. Kehl, Gang Wu, Hoon T. Chung, Michael Y. Hu, E. Ercan Alp, Karren L. More, Plotr Zelenay, Edward F. Holby, and Michael L. Neidig
J. Phys. Chem. C 2017, 121, 16283-16290
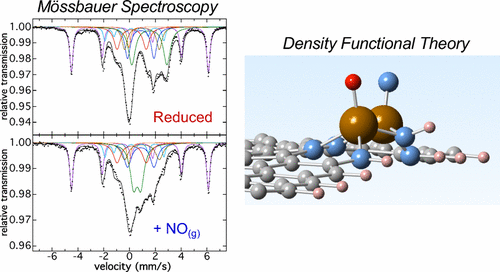
Nonprecious metal M–N–C (M = Fe or Co) catalysts that are effective for the oxygen-reduction reaction in polymer-electrolyte fuel cells have been developed, but no consensus has yet been reached regarding the nature of the M sites in these heterogeneous catalysts that are responsible for the reaction with dioxygen (O2). While multiple studies have developed correlations between Fe distributions in as-prepared catalysts and ORR activity, the direct identification of sites reactive toward O2 or O2-analogue molecules remains a significant challenge. In the present study, we demonstrate a new approach to identifying and characterizing potential Fe active sites in complex ORR catalysts that combines an effective probe molecule (NO(g)), Mössbauer spectroscopy, and nuclear resonance vibrational spectroscopy (NRVS) with density functional theory (DFT) calculations. Mössbauer spectroscopic studies demonstrate that NO(g) treatment of electrochemically reduced PANI–57Fe–C leads to a selective reaction with only a subset of the Fe species present. Nuclear resonance vibrational spectroscopic studies identified new Fe–ligand vibrations associated with the site reactive toward NO(g). DFT calculations of the vibrational properties of a selection of previously proposed active-site structures suggest that graphene zigzag edge-hosted Fe–N structures may be responsible for the observed vibrational behavior with NO(g) probe molecules. Furthermore, such sites are likely also reactive to O2, possibly serving as the ORR active sites in the synthesized materials.
Feng Li, Stephanie H. Carpenter, Robert F. Higgins, Mark G. Hitt, William W. Brennessel, Maryline G. Ferrier, Samantha K. Cary, Juan S. Lezama-Pacheco, Joshua T. Wright, Benjamin W. Stein, Matthew P. Shores, Michael L. Neidig, Stosh A. Kozimor, and Ellen M. Matson
Inorg. Chem. 2017, 56, 7065-7080
Nonprecious metal M–N–C (M = Fe or Co) catalysts that are effective for the oxygen-reduction reaction in polymer-electrolyte fuel cells have been developed, but no consensus has yet been reached regarding the nature of the M sites in these heterogeneous catalysts that are responsible for the reaction with dioxygen (O2). While multiple studies have developed correlations between Fe distributions in as-prepared catalysts and ORR activity, the direct identification of sites reactive toward O2 or O2-analogue molecules remains a significant challenge. In the present study, we demonstrate a new approach to identifying and characterizing potential Fe active sites in complex ORR catalysts that combines an effective probe molecule (NO(g)), Mössbauer spectroscopy, and nuclear resonance vibrational spectroscopy (NRVS) with density functional theory (DFT) calculations. Mössbauer spectroscopic studies demonstrate that NO(g) treatment of electrochemically reduced PANI–57Fe–C leads to a selective reaction with only a subset of the Fe species present. Nuclear resonance vibrational spectroscopic studies identified new Fe–ligand vibrations associated with the site reactive toward NO(g). DFT calculations of the vibrational properties of a selection of previously proposed active-site structures suggest that graphene zigzag edge-hosted Fe–N structures may be responsible for the observed vibrational behavior with NO(g) probe molecules. Furthermore, such sites are likely also reactive to O2, possibly serving as the ORR active sites in the synthesized materials.
Frédéric Gendron, Valerie E. Fleischauer, Thomas J. Duignan, Brian L. Scott, Matthias W. Löble, Smantha K. Cary, Stosh A. Kozimor, Hélène Bolvin, Michael L. Neidig, and Jochen Autschbach
Phys. Chem. Chem. Phys. 2017, 19, 17300-17313
We present a combined ab initio theoretical and experimental study of the magnetic circular dichroism (MCD) spectrum of the octahedral UCl6− complex ion in the UV-Vis spectral region. The ground state is an orbitally non-degenerate doublet E5/2u and the MCD is a C-term spectrum caused by spin–orbit coupling. Calculations of the electronic spectrum at various levels of theory indicate that differential dynamic electron correlation has a strong influence on the energies of the dipole-allowed transitions and the envelope of the MCD spectrum. The experimentally observed bands are assigned to dipole-allowed ligand-to-metal charge transfer into the 5f shell, and 5f to 6d transitions. Charge transfer excitations into the U 6d shell appear at much higher energies. The MCD-allowed transitions can be assigned via their signs of the C-terms: Under Oh double group symmetry, E5/2u → E5/2g transitions have negative C-terms whereas E5/2u → F3/2g transitions have positive Image C-terms if the ground state g-factor is negative, as it is the case for UCl6−.
Jared L. Kneebone, William W. Brennessel, and Michael L. Neidig
J. Am. Chem. Soc. 2017, 139, 6988-7003
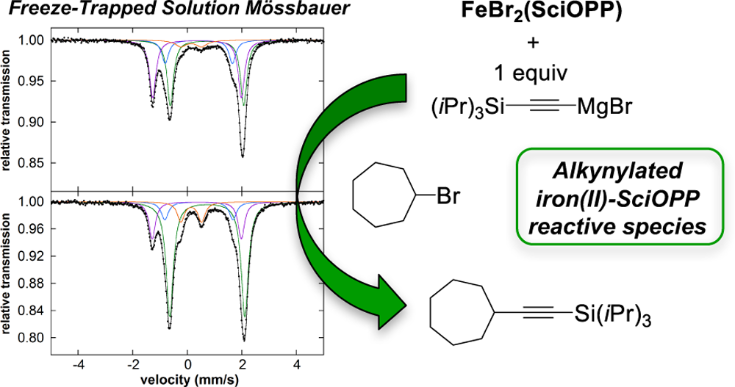
Iron-catalyzed cross-coupling reactions using alkynyl nucleophiles represent an attractive approach for the incorporation of alkynyl moieties into organic molecules. In the present study, a multitechnique approach combining inorganic spectroscopic methods, inorganic synthesis, and reaction studies is applied to iron-SciOPP catalyzed alkynyl-alkyl cross-couplings, providing the first detailed insight into the effects of variation from sp2- to sp-hybridized nucleophiles on iron speciation and reactivity. Reaction studies demonstrate that reaction of FeBr2(SciOPP) with 1 equiv (triisopropylsilyl)ethynylmagnesium bromide (TIPS-CC-MgBr) leads to a distribution of mono-, bis-, and tris-alkynylated iron(II)-SciOPP species due to rapid alkynyl ligand redistribution. While solvents such as THF promote these complex redistribution pathways, nonpolar solvents such as toluene enable increased stabilization of these iron species and further enabled assessment of their reactivity with electrophile. While the tris-alkynylated iron(II)-SciOPP species was found to be unreactive with the cycloheptyl bromide electrophile over the average turnover time of catalysis, the in situ formed neutral mono- and bis-alkynylated iron(II)-SciOPP complexes are consumed upon reaction with the electrophile with concomitant generation of cross-coupled product at catalytically relevant rates, indicating the ability of one or both of these species to react selectively with the electrophile. The nature of the reaction solvent and Grignard reagent addition rate were found to have broader implications in overall reaction selectivity, reaction rate, and accessibility of off-cycle iron(I)-SciOPP species. Additionally, the effects of steric substitution of the alkynyl Grignard reagent on catalytic performance were investigated. Fundamental insight into iron speciation and reactivity with alkynyl nucleophiles reported herein provides an essential foundation for the continued development of this important class of reactions.
Matthew C. Leclerc, Bulat M. Gabidullin, Jason G. Da Gama, Stephanie L. Daifuku, Theresa E. Iannuzzi, Michael L. Neidig, and R. Tom Baker
Organometallics 2017, 36, 849-857
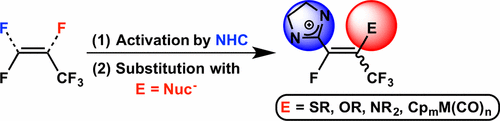
Herein, a recently reported polyfluoroalkenyl imidazolium salt is shown to react with nitrogen-, oxygen- and sulfur-based nucleophiles at the Cβ position in a stereoselective and regioselective fashion, without the use of a transition metal. In contrast, reactivity with 1-methylimidazole demonstrates net substitution at Cα. This product reacts quantitatively with water, affording clean transformation of a difluoromethylene group to give an α,β-unsaturated trifluoromethyl ketone. Further reactivity studies demonstrate that the difluoromethyl fragment of an N-heterocyclic fluoroalkene is capable of direct C–C bond formation with NaCp through loss of sodium fluoride and double C–F bond activation (Cp = cyclopentadienide). TD-DFT calculations of this product indicate that both the HOMO and LUMO are of mixed π/π* character and are delocalized over the N-heterocyclic and Cp fragments, giving rise to a very intense absorption feature in the UV–vis spectrum. Additionally, two carbonylmetalate-substituted fluorovinyl imidazolium complexes featuring Mn and Mo were isolated and fully characterized.
Tessa M. Baker, Toshiki G. Nakashige, Elizabeth M. Nolan, and Michael L. Neidig
Chem. Sci. 2017, 8, 1369-1377
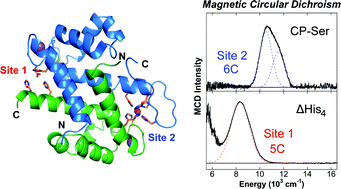
Calprotectin (CP) is an abundant metal-chelating protein involved in host defense, and the ability of human CP to bind Fe(II) in a calcium-dependent manner was recently discovered. In the present study, near-infrared magnetic circular dichroism spectroscopy is employed to investigate the nature of Fe(II) coordination at the two transition-metal-binding sites of CP that are a His3Asp motif (site 1) and a His6 motif (site 2). Upon the addition of sub-stoichiometric Fe(II), a six-coordinate (6C) Fe(II) center associated with site 2 is preferentially formed in the presence of excess Ca(II). This site exhibits an exceptionally large ligand field (10Dq = 11 045 cm−1) for a non-heme Fe(II) protein. Analysis of CP variants lacking residues of the His6 motif supports that CP coordinates Fe(II) at site 2 by employing six His ligands. In the presence of greater than one equiv. of Fe(II) or upon mutation of the His6 motif, the metal ion also binds at site 1 of CP to form a five-coordinate (5C) Fe(II)–His3Asp motif that was previously unidentified in this system. Notably, the introduction of His-to-Ala mutations at the His6 motif results in a mixture of 6C (site 2) and 5C (site 1) signals in the presence of sub-stoichiometric Fe(II). These results are consistent with a reduced Fe(II)-binding affinity of site 2 as more weakly coordinating water-derived ligands complete the 6C site. In the absence of Ca(II), both sites 1 and 2 are occupied upon addition of sub-stoichiometric Fe(II), and a stronger ligand field is observed for the 5C site. These spectroscopic studies provide further evaluation of a unique non-heme Fe(II)–His6 site for metalloproteins and support the notion that Ca(II) ions influence the Fe(II)-binding properties of CP.
2016
Magnetic Circular Dichroism and Density Functional Theory Studies of Iron(II)-Pincer Complexes: Insight into Electronic Structure and Bonding Effects of Pincer N-Heterocyclic Carbene Moieties
Catalytic Light-Driven Generation of Hydrogen from Water by Iron Dithiolene Complexes
Manipulating Magneto-Optic Properties of a Chiral Polymer by Doping with Stable Organic Biradicals
Facile hydrogen atom transfer to iron(iii) imido radical complexes supported by a dianionic pentadentate ligand
Resident holes and electrons at organic/conductor and organic/organic interfaces: An electron paramagnetic resonance investigation
Isolation, Characterization, and Reactivity of Fe8Me12–: Kochi’s S = 1/2 Species in Iron-Catalyzed Cross-Couplings with MeMgBr and Ferric Salts
Mononuclear, Dinuclear, and Trinuclear Iron Complexes Featuring a New Monoanionic SNS Thiolate Ligand
Electronic Structure and Bonding in Iron(II) and Iron(I) Complexes Bearing Bisphosphine Ligands of Relevance to Iron-Catalyzed C–C Cross-Coupling
Tessa M. Baker, Teresa L. Mako, Aristidis Vasilopoulos, Bo Li, Jeffery A. Byers, and Michael L. Neidig
Organometallics 2016, 35, 3692–3700
Iron complexes containing pincer ligands that incorporate N-heterocyclic carbene (NHC) moieties are of significant interest in organometallic catalysis in order to generate more oxidatively robust complexes that may exhibit novel catalytic properties. In order to define the effect that introducing NHC moieties into pincer ligands has on electronic structure and bonding in iron(II)-pincer complexes, MCD and DFT studies of (iPrCDA)FeBr2, (iPrPDI)FeBr2, and (iPrCNC)FeBr2were performed. These studies quantify the electronic structures and bonding interactions as a function of pincer ligand variation. They also demonstrate that the observed ligand fields (and, hence, spin states) directly correlate to the increased Fe–C bonding and pincer-donating abilities that result from introducing NHC moieties into the pincer ligand. However, the net donor abilities of the pincers and the strength of the Fe-pincer interaction do not directly correlate to the number of NHC moieties present, but instead are determined to be due to differences in Fe–C and overall Fe-pincer bonding as a result of the position of the NHC moieties in the pincer ligand and the overall geometric constraints of the pincer architecture.
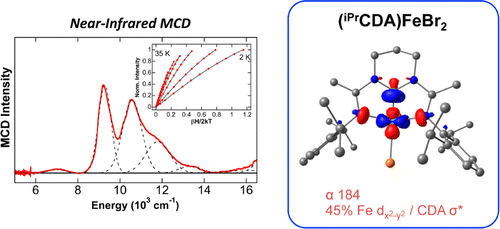
Hongjin Lv, T. Purnima A. Ruberu, Valerie E. Fleischauer, William W. Brennessel, Michael L. Neidig, and Richard Eisenberg
J. Am. Chem. Soc. 2016, 138, 11654–11663
The development of active, robust systems for light-driven hydrogen production from aqueous protons based on catalysts and light absorbers composed solely of earth abundant elements remains a challenge in the development of an artificial photosynthetic system for water splitting. Herein, we report the synthesis and characterization of four closely related Fe bis(benzenedithiolate) complexes that exhibit catalytic activity for hydrogen evolution when employed in systems with water-soluble CdSe QDs as photosensitizer and ascorbic acid as a sacrificial electron source under visible light irradiation (520 nm). The complex with the most electron-donating dithiolene ligand exhibits the highest activity, the overall order of activity correlating with the reduction potential of the formally Fe(III) dimeric dianions. Detailed studies of the effect of different capping agents and the extent of surface coverage of these capping agents on the CdSe QD surfaces reveal that they affect system activity and provide insight into the continued development of such systems containing QD light absorbers and molecular catalysts for H2 formation.
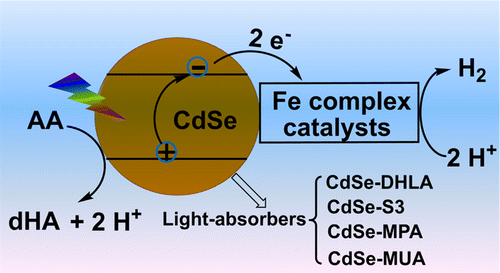
Chang-Keun Lim, Min Ju Cho, Ajay Singh, Qi Li, Won Jin Kim, Hong Sub Jee, Kathlyn L. Fillman, Stephanie H. Carpenter, Michael L. Neidig, Alexander Baev, Mark T. Swihart, and Paras N. Prasad
Nano Lett. 2016, 16, 5451–5455
The development of active, robust systems for light-driven hydrogen production from aqueous protons based on catalysts and light absorbers composed solely of earth abundant elements remains a challenge in the development of an artificial photosynthetic system for water splitting. Herein, we report the synthesis and characterization of four closely related Fe bis(benzenedithiolate) complexes that exhibit catalytic activity for hydrogen evolution when employed in systems with water-soluble CdSe QDs as photosensitizer and ascorbic acid as a sacrificial electron source under visible light irradiation (520 nm). The complex with the most electron-donating dithiolene ligand exhibits the highest activity, the overall order of activity correlating with the reduction potential of the formally Fe(III) dimeric dianions. Detailed studies of the effect of different capping agents and the extent of surface coverage of these capping agents on the CdSe QD surfaces reveal that they affect system activity and provide insight into the continued development of such systems containing QD light absorbers and molecular catalysts for H2 formation.
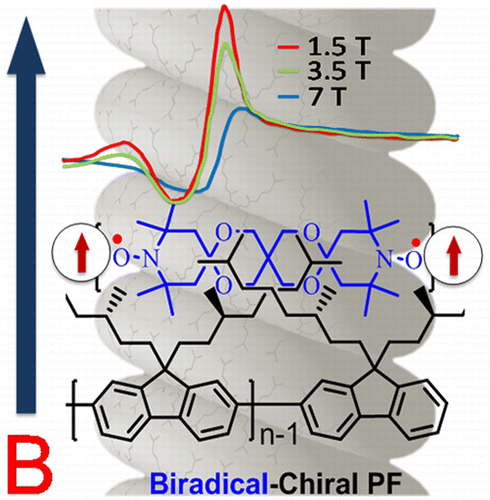
Denis M. Spasyuk, Stephanie H. Carpenter, Christos E. Kefalidis, Warren E. Piers, Michael L. Neidig, and Laurent Maron
Chem. Sci. 2016, 7, 5939-5944
A dianionic tetrapodal pentadentate diborate ligand is introduced. This ligand forms a high spin neutral iron(II) complex that reacts with a variety of organoazides to yield transient Fe(III) imido radicals that are extremely potent hydrogen atom abstractors. The nature of these species is supported by full characterization of the Fe(III) amido products, kinetic studies, density functional computations and Mössbauer spectroscopy on the –C6H4-p-tBu substituted derivative.
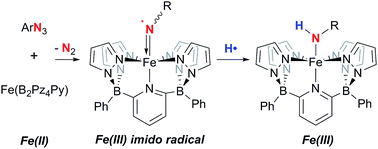
Chen Zhang, Stephanie L. Daifuku, Michael L. Neidig, and Alfred P. Marchetti
Org. Electron. 2016, 37, 379-385
A number of organic/conductor and organic/organic interfaces have been examined by EPR spectroscopy to ascertain the areal concentration of organic ions at the interface. Organic hole transport materials such as NPB and TAPC at an interface with MoOx are found to have areal concentrations on the order of 1014 cations per cm2. C60 at an interface with MoOx creates ≈1013 cations per cm2 depending on the roughness of the substrate. However, C60 at an interface with Mg or Ag produces only about 4 × 1012 anions per cm2. Ion concentrations are generally in accord with the energy levels (adiabatic IP, EA etc) of the two materials at a given interface.
Salvador B. Muñoz III, Stephanie L. Daifuku, William W. Brennessel, and Michael L. Neidig
J. Am. Chem. Soc. 2016, 138, 7492-7495
Iron-catalyzed cross-couplings with simple ferric salts have been known since the 1970s, pioneered by Kochi for cross-coupling using alkylmagnesium nucleophiles including MeMgBr. While Kochi observed the formation of a S = 1/2 iron species in reactions of simple ferric salts with MeMgBr proposed to be an iron(I) species, the identity of this species has remained undefined for nearly 40 years. Herein, we report the isolation and characterization of [MgCl(THF)5][Fe8Me12], which combined with EPR and MCD studies is shown to be consistent with Kochi’s S = 1/2 species. Reaction studies with β-bromostyrene demonstrate that this species alone displays minimal reactivity but, when combined with additional MeMgBr, leads to rapid and selective formation of cross-coupled product.
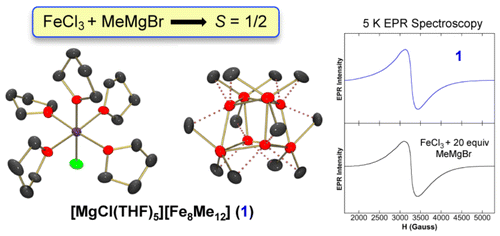
Uttam K. Das, Stepahnie L. Daifuku, Serge I. Gorelsky, Ilia Korobkov, Michael L. Neidig, Jennifer J. Le Roy, Muralee Murugesu, and R. Tom Baker
Inorg. Chem. 2016, 55, 987-997
The new tridentate ligand, SMeNHS = 2-(2-methylthiophenyl)benzothiazolidine, prepared in a single step from commercial precursors in excellent yield, undergoes ring-opening on treatment with Fe(OTf)2 in the presence of base affording a trinuclear iron complex, [Fe3(μ2-SMeNS–)4](OTf)2 (1) which is fully characterized by structural and spectroscopic methods. X-ray structural data reveal that 1 contains four SMeNS– ligands meridionally bound to two pseudooctahedral iron centers each bridged by two thiolates to a distorted tetrahedral central iron. The combined spectroscopic (UV–vis, Mössbauer, NMR), magnetic (solution and solid state), and computational (DFT) studies indicate that 1 includes a central, high-spin Fe(II) (S = 2) with two low-spin (S = 0) peripheral Fe(II) centers. Complex 1 reacts with excess PMePh2, CNxylyl (2,6-dimethylphenyl isocyanide), and P(OMe)3 in CH3CN to form diamagnetic, thiolate-bridged, dinuclear Fe(II) complexes {[Fe(μ-SMeNS–)L2]2}(OTf)2 (2–4). These complexes are characterized by elemental analysis; 1H NMR, IR, UV–vis, and Mössbauer spectroscopy; and single crystal X-ray diffraction. Interestingly, addition of excess P(OMe)3 to complex 1 in CH2Cl2 produces primarily the diamagnetic, mononuclear Fe(II) complex, {Fe(SMeNS–)[P(OMe)3]3}(OTf) (5).
Jared L. Kneebone, Valerie E. Fleischauer, Stephanie L. Daifuku, Ari A. Shaps, Joseph M. Bailey, Theresa E. Iannuzzi, and Michael L. Neidig
Inorg. Chem. 2016, 55, 272-282
Chelating phosphines are effective additives and supporting ligands for a wide array of iron-catalyzed cross-coupling reactions. While recent studies have begun to unravel the nature of the in situ-formed iron species in several of these reactions, including the identification of the active iron species, insight into the origin of the differential effectiveness of bisphosphine ligands in catalysis as a function of their backbone and peripheral steric structures remains elusive. Herein, we report a spectroscopic and computational investigation of well-defined FeCl2(bisphosphine) complexes (bisphosphine = SciOPP, dpbz, tBudppe, or Xantphos) and known iron(I) variants to systematically discern the relative effects of bisphosphine backbone character and steric substitution on the overall electronic structure and bonding within their iron complexes across oxidation states implicated to be relevant in catalysis. Magnetic circular dichroism (MCD) and density functional theory (DFT) studies demonstrate that common o-phenylene and saturated ethyl backbone motifs result in small but non-negligible perturbations to 10Dq(Td) and iron–bisphosphine bonding character at the iron(II) level within isostructural tetrahedra as well as in five-coordinate iron(I) complexes FeCl(dpbz)2 and FeCl(dppe)2. Notably, coordination of Xantphos to FeCl2 results in a ligand field significantly reduced relative to those of its iron(II) partners, where a large bite angle and consequent reduced iron–phosphorus Mayer bond orders (MBOs) could play a role in fostering the unique ability of Xantphos to be an effective additive in Kumada and Suzuki–Miyaura alkyl–alkyl cross-couplings. Furthermore, it has been found that the peripheral steric bulk of the SciOPP ligand does little to perturb the electronic structure of FeCl2(SciOPP) relative to that of the analogous FeCl2(dpbz) complex, potentially suggesting that differences in the steric properties of these ligands might be more important in determining in situ iron speciation and reactivity.
2015
Crystal structure of a third polymorph of tris(acetylacetonato-κ2O,O′)iron(III)
Possible Demonstration of a Polaronic Bose-Einstein(-Mott) Condensate in UO2(+x) by Ultrafast THz Spectroscopy and Microwave Dissipation
Iron(II) Active Species in Iron–Bisphosphine Catalyzed Kumada and Suzuki–Miyaura Cross-Couplings of Phenyl Nucleophiles and Secondary Alkyl Halides
Linear and T-Shaped Iron(I) Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis and Structure Characterization
Ambivalent binding between a radical-based pincer ligand and iron
How Innocent are Potentially Redox Non-Innocent Ligands? Electronic Structure and Metal Oxidation States in Iron-PNN Complexes as a Representative Case Study
A combined magnetic circular dichroism and density functional theory approach for the elucidation of electronic structure and bonding in three- and four-coordinate iron(ii)–N-heterocyclic carbene complexes
Tessa M. Baker, Kevin M. Howard, William W. Brennessel, and Michael L. Neidig
Acta Cryst. 2015, E71, m228-m229
In the structure of the title complex, [Fe(C5H7O2)3] or Fe(acac)3, the asymmetric unit contains one molecule in a general position. The coordination sphere of the FeIII atom is that of a slightly distorted octahedron. The crystal under investigation was a two-component pseudo-merohedral twin in the monoclinic system with a β angle close to 90°. Twin law [100/0-10/00-1] reduced the R1 residual [I > 2σ(I)] from 0.0769 to 0.0312, and the mass ratio of twin components refined to 0.8913 (5):0.1087 (5). In the crystal, molecules are arranged in sheets normal to [001] via non-classical C—H⋯O hydrogen bonding. No other significant intermolecular interactions are observed. The structure is a new polymorph of Fe(acac)3 and is isotypic with one polymorph of its gallium analog.
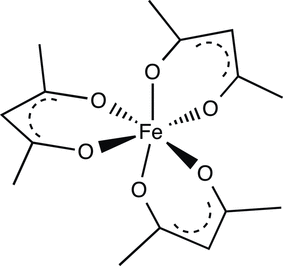
Steven D. Conradson, Steven M.Gilbertson, Stephanie L. Daifuku, Jeffery A. Kehl, Tomasz Durakiewicz, David A. Anderson, Alan R. Bishop, Darrin D. Byler, Pablo Maldonado, Peter M. Oppeneer, James A. Valdez, Michael L. Neidig, and George Rodirguez
Scientific Reports 2015, 5, 15278
Bose-Einstein condensates (BECs) composed of polarons would be an advance because they would combine coherently charge, spin and a crystal lattice. Following our earlier report of unique structural and spectroscopic properties, we now identify potentially definitive evidence for polaronic BECs in photo- and chemically doped UO2(+x) on the basis of exceptional coherence in the ultrafast time dependent terahertz absorption and microwave spectroscopy results that show collective behavior including dissipation patterns whose precedents are condensate vortex and defect disorder and condensate excitations. That some of these signatures of coherence in an atom-based system extend to ambient temperature suggests a novel mechanism that could be a synchronized, dynamical, disproportionation excitation, possibly via the solid state analog of a Feshbach resonance that promotes the coherence. Such a mechanism would demonstrate that the use of ultra-low temperatures to establish the BEC energy distribution is a convenience rather than a necessity, with the actual requirement for the particles being in the same state that is not necessarily the ground state attainable by other means. A macroscopic quantum object created by chemical doping that can persist to ambient temperature and resides in a bulk solid would be revolutionary in a number of scientific and technological fields.
Stephanie L. Daifuku, Jared L. Kneebone, Benjamin E. R. Snyder, and Michael L. Neidig
J. Am. Chem. Soc. 2015, 137, 11432-11444
While previous studies have identified FeMes2(SciOPP) as the active catalyst species in iron–SciOPP catalyzed Kumada cross-coupling of mesitylmagnesium bromide and primary alkyl halides, the active catalyst species in cross-couplings with phenyl nucleophiles, where low valent iron species might be prevalent due to accessible reductive elimination pathways, remains undefined. In the present study, in situ Mössbauer and magnetic circular dichroism spectroscopic studies combined with inorganic syntheses and reaction studies are employed to evaluate the in situ formed iron species and identify the active catalytic species in iron–SciOPP catalyzed Suzuki–Miyaura and Kumada cross-couplings of phenyl nucleophiles and secondary alkyl halides. While reductive elimination to form Fe(η6-biphenyl)(SciOPP) occurs upon reaction of FeCl2(SciOPP) with phenyl nucleophiles, this iron(0) species is not found to be kinetically competent for catalysis. Importantly, mono- and bis-phenylated iron(II)–SciOPP species that form prior to reductive elimination are identified, where both species are found to be reactive toward electrophile at catalytically relevant rates. The higher selectivity toward the formation of cross-coupled product observed for the monophenylated species combined with the undertransmetalated nature of the in situ iron species in both Kumada and Suzuki–Miyaura reactions indicates that Fe(Ph)X(SciOPP) (X = Br, Cl) is the predominant reactive species in cross-coupling. Overall, these studies demonstrate that low-valent iron is not required for the generation of highly reactive species for effective aryl-alkyl cross-couplings.
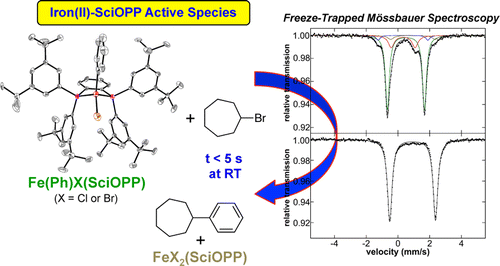
Zhenwu Ouyang, Jingzhen Du, Lei Wang, Jared L. Kneebone, Michael L. Neidig, and Liang Deng
Inorg. Chem. 2015, 54, 8808-8816
The use of the N-heterocyclic carbene (NHC) ligands 1,3-bis(2′,6′-diethylphenyl)-4,5-(CH2)4-imidazol-2-ylidene (cyIDep), 1,3-bis(2′,6′-diethylphenyl)-imidazolin-2-ylidene (sIDep), and its N-mesityl analogue sIMes enables the preparation of the two-coordinate homoleptic iron(I)-NHC complexes [(cyIDep)2Fe][BArF4] (3, ArF denoted for 3,5-di(trifluoromethyl)phenyl) and [(sIDep)2Fe][BArF4] (4) and the T-shaped iron(I)-NHC complex [(sIMes)2Fe(THF)][BPh4] (5, THF = tetrahydrofuran). Complexes 3–5 were prepared via the sequential protocol of control reduction of iron(II) dihalides by KC8 in the presence of the corresponding NHC ligands followed by halide-abstraction with NaBAr4. Spectroscopic characterization, including single-crystal X-ray diffraction studies and 57Fe Mössbauer spectroscopy, in combination with density functional theory calculations, suggest their high-spin nature. Solution property study (absorption spectroscopy and cyclic voltammetry) indicates that 3 and 5 keep their corresponding two- and three-coordinate nature in THF solution, and 4 might reversibly coordinate a THF molecule to form, presumably, the T-shaped species [(sIDep)2Fe(THF)][BArF4]. The isolation of 3 and 4 demonstrates the accessibility of homoleptic two-coordinate iron(I)-NHC complexes.
Katie L. M. Harriman, Alicea A. Leitch, Sebastian A. Stoian, Fatemah Habib, Jared L. Kneebone, Serge I. Gorelsky, Ilia Korobkov, Serge Desgreniers, Michael L. Neidig, Stephen Hill, Muralee Murugesu, and Jaclyn L. Brusso
Dalton Trans. 2015, 44, 10516-10523
A complex exhibiting valence delocalization was prepared from 3,5-bis(2-pyridyl)-1,2,4,6-thiatriazinyl (Py2TTA˙), an inherently redox active pincer-type ligand, coordinated to iron (Fe(Py2TTA)Cl2 (1)). Complex 1 can be prepared via two routes, either from the reaction of the neutral radical with FeCl2 or by treatment of the anionic ligand (Py2TTA−) with FeCl3, demonstrating its unique redox behaviour. Electrochemical studies, solution absorption and solid-state diffuse reflectance measurements along with X-ray crystallography were carried out to elucidate the molecular and solid-state properties. Temperature- and field-dependent Mössbauer spectroscopy coupled with magnetic measurements revealed that 1 exhibits an isolated S = 5/2 ground spin state for which the low-temperature magnetic behaviour is dominated by exchange interactions between neighbouring molecules. This ground state is rationalized on the basis of DFT calculations that predict the presence of strong electronic interactions between the redox active ligand and metal. This interaction leads to the delocalization of β electron density over the two redox active centres and highlights the difficulty in assigning formal charges to 1.
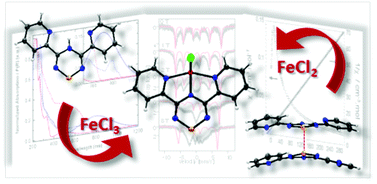
Burkhard Butschke, Kathlyn L. Fillman, Tatyana Bendikov, Linda J. W. Shimon, Yael Diskin-Posner, Gregory Leitus, Serge I. Gorelsky, Michael L. Neidig, and David Milstein
Inorg. Chem. 2015, 54, 4909-4926
Herein we present a series of new α-iminopyridine-based iron-PNN pincer complexes [FeBr2LPNN] (1), [Fe(CO)2LPNN] (2), [Fe(CO)2LPNN](BF4) (3), [Fe(F)(CO)2LPNN](BF4) (4), and [Fe(H)(CO)2LPNN](BF4) (5) with formal oxidation states ranging from Fe(0) to Fe(II) (LPNN = 2-[(di-tert-butylphosphino)methyl]-6-[1-(2,4,6-mesitylimino)ethyl]pyridine). The complexes were characterized by a variety of methods including 1H, 13C, 15N, and 31P NMR, IR, Mössbauer, and X-ray photoelectron spectroscopy (XPS) as well as electron paramagnetic resonance (EPR) and magnetic circular dichroism (MCD) spectroscopy, SQUID magnetometry, and X-ray crystallography, focusing on the assignment of the metal oxidation states. Ligand structural features suggest that the α-iminopyridine ligand behaves as a redox non-innocent ligand in some of these complexes, particularly in [Fe(CO)2LPNN] (2), in which it appears to adopt the monoanionic form. In addition, the NMR spectroscopic features (13C, 15N) indicate the accumulation of charge density on parts of the ligand for 2. However, a combination of spectroscopic measurements that more directly probe the iron oxidation state (e.g., XPS), density functional theory (DFT) calculations, and electronic absorption studies combined with time-dependent DFT calculations support the description of the metal atom in 2 as Fe(0). We conclude from our studies that ligand structural features, while useful in many assignments of ligand redox non-innocence, may not always accurately reflect the ligand charge state and, hence, the metal oxidation state. For complex 2, the ligand structural changes are interpreted in terms of strong back-donation from the metal center to the ligand as opposed to electron transfer.
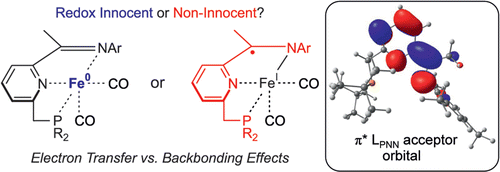
Kathlyn L. Fillman, Jacob A. Przyojski, Malik H. Al-Afyouni, Zachary J. Tonzetich, and Michael L. Neidig
Chem. Sci. 2015, 6, 1178-1188
Iron salts and N-heterocyclic carbene (NHC) ligands is a highly effective combination in catalysis, with observed catalytic activities being highly dependent on the nature of the NHC ligand. Detailed spectroscopic and electronic structure studies have been performed on both three- and four-coordinate iron(II)–NHC complexes using a combined magnetic circular dichroism (MCD) and density functional theory (DFT) approach that provide detailed insight into the relative ligation properties of NHCs compared to traditional phosphine and amine ligands as well as the effects of NHC backbone structural variations on iron(II)–NHC bonding. Near-infrared MCD studies indicate that 10Dq(Td) for (NHC)2FeCl2 complexes is intermediate between those for comparable amine and phosphine complexes, demonstrating that such iron(II)–NHC and iron(II)–phosphine complexes are not simply analogues of one another. Theoretical studies including charge decomposition analysis indicate that the NHC ligands are slightly stronger donor ligands than phosphines but also result in significant weakening of the Fe–Cl bonds compared to phosphine and amine ligands. The net result is significant differences in the d orbital energies in four-coordinate (NHC)2FeCl2 complexes relative to the comparable phosphine complexes, where such electronic structure differences are likely a significant contributing factor to the differing catalytic performances observed with these ligands. Furthermore, Mössbauer, MCD and DFT studies of the effects of NHC backbone structure variations (i.e. saturated, unsaturated, chlorinated) on iron–NHC bonding and electronic structure in both three- and four-coordinate iron(II)–NHC complexes indicate only small differences as a function of backbone structure, that are likely amplified at lower oxidation states of iron due to the resulting decrease in the energy separation between the occupied iron d orbitals and the unoccupied NHC π* orbitals.
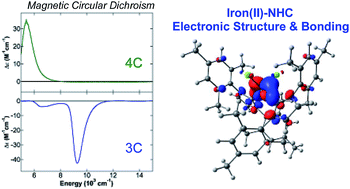
2014
Isolation and Characterization of a Tetramethyliron(III) Ferrate: An Intermediate in the Reduction Pathway of Ferric Salts with MeMgBr
Direct observation of ICT cations at the HTL/transparent semiconductor interface
Iron Phosphine Catalyzed Cross-Coupling of Tetraorganoborates and Related Group 13 Nucleophiles with Alkyl Halides
Two- and three-coordinate formal iron(i) compounds featuring monodentate aminocarbene ligands
A Combined Mössbauer, Magnetic Circular Dichroism, and Density Functional Theory Approach for Iron Cross-Coupling Catalysis: Electronic Structure, In Situ Formation, and Reactivity of Iron-Mesityl-Bisphosphines
Flexible Binding of PNP Pincer Ligands to Monomeric Iron Complexes
Iron Dicarbonyl Complexes Featuring Bipyridine‐Based PNN Pincer Ligands with Short Interpyridine C-C Bond Lengths: Innocent or Non‐Innocent Ligand?
Reactivity of (NHC)2FeX2 Complexes toward Arylborane Lewis Acids and Arylboronates
Malik H. Al-Afyouni, Kathlyn L. Fillman, William W. Brennessel, and Michael L. Neidig
J. Am. Chem. Soc. 2014, 136, 15457-15460
While iron-catalyzed Kumada cross-coupling reactions with simple iron salts have been known since the early 1970s, the nature of the in situ-formed iron species remains elusive. Herein, we report the synthesis of the homoleptic tetralkyliron(III) ferrate complex [MgCl(THF)5][FeMe4] from the reaction of FeCl3 with MeMgBr in THF. Upon warming, this distorted square-planar S = 3/2 species converts to the S = 1/2 species originally observed by Kochi and co-workers with concomitant formation of ethane, consistent with its intermediacy in the reduction pathway of FeCl3 to generate the reduced iron species involved in catalysis.
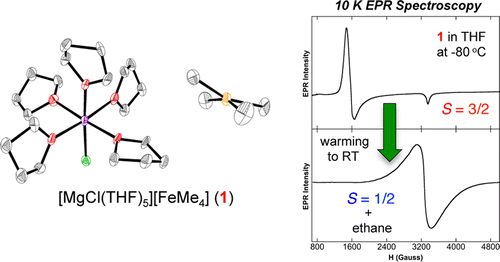
Stephanie L. Daifuku, Chris Favaro, Alfred P. Marchetti, and Michael L. Neidig
Org. Electron. 2014, 15, 3761-3765
The cation density at the interface of a transparent anode and an organic layer has been measured for several hole transport (HT) materials. The number of cations at the interface of ITO:MoOx with rubrene, NPB, m-MTDATA and TCTA was found to range from 8 × 1013 to 1.5 × 1014 per cm2 in freshly prepared devices. These values decreased by about 25% after one month. These cations are part of the dipole layer that results from the transfer of electrons from an organic layer, whose adiabatic ionization potential is less than the work function of the anode.
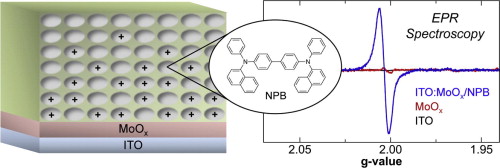
Robin B. Bedford, Peter B. Brenner, Emma Carter, Jamie Clifton, Paul M. Cogswell, Nicholas J. Gower, Mairi F. Haddow, Jeremy N. Harvey, Jeffrey A. Kehl, Damien M. Murphy, Emily C. Neeve, Michael L. Neidig, Joshua Nunn, Benjamin E. R. Synder, and Joseph Taylor
Organometallics 2014, 33, 5767-5780
Iron phosphine complexes prove to be good precatalysts for the cross-coupling of alkyl, benzyl, and allyl halides with not only aryl triorganoborate salts but also related aluminum-, gallium-, indium-, and thallium-based nucleophiles. Mechanistic studies revealed that while Fe(I) can be accessed on catalytically relevant time scales, lower average oxidation states are not formed fast enough to be relevant to catalysis. EPR spectroscopic studies reveal the presence of bis(diphosphine)iron(I) complexes in representative catalytic reactions and related processes with a range of group 13 nucleophiles. Isolated examples were studied by Mössbauer spectroscopy and single-crystal X-ray structural analysis, while the electronic structure was probed by dispersion-corrected B3LYP DFT calculations. An EPR study on an iron system with a bulky diphosphine ligand revealed the presence of an S = 1/2 species consistent with the formation of a mono(diphosphine)iron(I) species with inequivalent phosphine donor environments. DFT analysis of model complexes allowed us to rule out a T-shaped Fe(I) structure, as this is predicted to be high spin.
Zhenbo Mo, Zhenwu Ouyang, Lei Wang, Kathlyn L. Fillman, Michael L. Neidig, and Liang Deng
Org. Chem. Front. 2014, 1, 1040-1044
Bulky monodentate aminocarbene ligands, IMes and Me2-cAAC (IMes: 1,3-bis(2′,4′,6′-trimethylphenyl)imidazol-2-ylidene; Me2-cAAC: 3,3,5,5-tetramethyl-1-(2′,6′-diisopropylphenyl)pyrrolidine-2-ylidene), have been shown to be effective in supporting formal 13- and 11-electron iron(I) species. From the reactions of ferrous precursors and one equivalent of a reducing agent, three-coordinate complexes of the type [L2FeCl] (L = IMes or Me2-cAAC) have been synthesized in good yields. A mixed-ligand complex [(IMes)(Me2-cAAC)FeCl] was prepared from the ligand substitution reaction of [(IMes)2FeCl] with Me2-cAAC. All of the three-coordinate iron complexes can react with Na[BArF]4, from which a two-coordinate species [(Me2-cAAC)2Fe][BArF4] has been isolated. Single-crystal X-ray diffraction studies established their molecular structures to be the first examples of two- and three-coordinate formal iron(I) species supported by carbene ligands. The large solution magnetic moments, differentiated Fe–C(carbene) distances and 57Fe Mössbauer isomer shifts are indicative of their rich electronic properties.
Stephanie L. Daifuku, Malik H. Al-Afyouni, Benjamin E. R. Synder, Jared L. Kneebone, and Michael L. Neidig
J. Am. Chem. Soc. 2014, 136, 9132-9143
While iron-bisphosphines have emerged as effective catalysts for C–C cross-coupling, the nature of the in situ formed iron species, elucidation of the active catalysts and the mechanisms of catalysis have remained elusive. A combination of 57Fe Mössbauer and magnetic circular dichroism (MCD) spectroscopies of well-defined and in situ formed mesityl-iron(II)-SciOPP species combined with density functional theory (DFT) investigations provides the first direct insight into electronic structure, bonding and in situ speciation of mesityl-iron(II)-bisphosphines in the Kumada cross-coupling of MesMgBr and primary alkyl halides using FeCl2(SciOPP). Combined with freeze-trapped solution Mössbauer studies of reactions with primary alkyl halides, these studies demonstrate that distorted square-planar FeMes2(SciOPP) is the active catalyst for cross-coupling and provide insight into the molecular-level mechanism of catalysis. These studies also define the effects of key reaction protocol details, including the role of the slow Grignard addition method and the addition of excess SciOPP ligand, in leading to high product yields and selectivities.
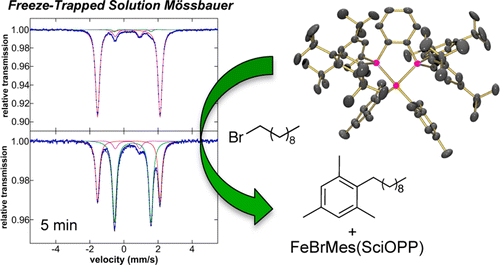
Kathlyn L. Fillman, Elizabeth A. Bielinski, Timothy J. Schmeier, Jared C. Nesvet, Tessa M. Woodruff, Cassie J. Pan, Michael K. Takase, Nilay Hazari, and Michael L. Neidig
Inorg. Chem. 2014, 53, 6066-6072
Transition metal complexes supported by pincer ligands have many important applications. Here, the syntheses of five-coordinate PNP pincer-supported Fe complexes of the type (PNP)FeCl2 (PNP = HN{CH2CH2(PR2)}2, R = iPr (iPrPNP), tBu (tBuPNP), or cyclohexyl (CyPNP)) are reported. In the solid state, (iPrPNP)FeCl2 was characterized in two different geometries by X-ray crystallography. In one form, the iPrPNP ligand binds to the Fe center in the typical meridional geometry for a pincer ligand, whereas in the other form, the iPrPNP ligand binds in a facial geometry. The electronic structures and geometries of all of the (PNP)FeCl2 complexes were further explored using 57Fe Mössbauer and magnetic circular dichroism spectroscopy. These measurements show that in some cases two isomers of the (PNP)FeCl2 complexes are present in solution and conclusively demonstrate that binding of the PNP ligand is flexible, which may have implications for the reactivity of this important class of compounds.
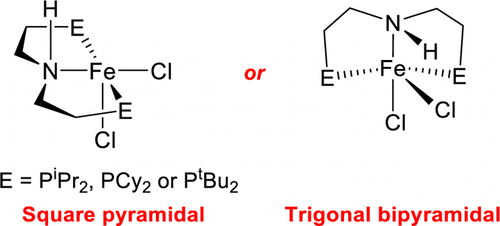
Thomas Zell, Petr Milko, Kathlyn L. Fillman, Yael Diskin-Posner, Tatyana Bendikov, Mark A. Iron, Gregory Leitus, Yehoshoa Ben-David, Michael L. Neidig, and David Milstein
Chem. Eur. J. 2014, 20, 4403-4413
A series of iron dicarbonyl complexes with bipyridine‐based PNN pincer ligands were synthesized and characterized by multinuclear NMR spectroscopy (1H, 13C, 15N, 31P), IR spectroscopy, cyclic voltammetry, 57Fe Mössbauer spectroscopy, XPS spectroscopy, and single‐crystal X‐ray diffraction. The complexes with the general formula [(R‐PNN)Fe(CO)2] (5 : R‐PNN=t Bu‐PNN=6‐[(di‐tert ‐butylphosphino)methyl]‐2,2′‐bipyridine, 6 : R‐PNN=i Pr‐PNN=6‐[(diisopropylphosphino)methyl]‐2,2′‐bipyridine, and 7 : R‐PNN=Ph‐PNN=6‐[(diphenylphosphino)methyl]‐2,2′‐bipyridine) feature differently P‐substituted PNN pincer ligands. Complexes 5 and 6 were obtained by reduction of the corresponding dihalide complexes [(R‐PNN)Fe(X)2] (1 : R=t Bu, X=Cl; 2 : R=t Bu, X=Br; 3 : R=i Pr, X=Cl; 4 : R=i Pr, X=Br) in the presence of CO. The analogous Ph‐substituted complex 7 was synthesized by a reaction of the free ligand with iron pentacarbonyl. The low‐spin complexes 5 –7 (S =0) are diamagnetic and have distorted trigonal bipyramidal structures in solution, whereas in the solid state the geometries around the iron are best described as distorted square pyramidal. Compared to other structurally characterized complexes with these PNN ligands, shortened interpyridine C-C bonds of about 1.43 Å were measured. A comparison with known examples, theoretically described as metal complexes bearing bipyridine π‐radical anions (bpy. −), suggests that the complexes can be described as FeI complexes with one electron antiferromagnetically coupled to the ligand‐based radical anions. However, computational studies, at the NEVPT2/CASSCF level of theory, reveal that the shortening of the C-C bond is a result of extensive π‐backbonding of the iron center into the antibonding orbital of the bpy unit. Hence, the description of the complexes as Fe0 complexes with neutral bipyridine units is the favorable one.
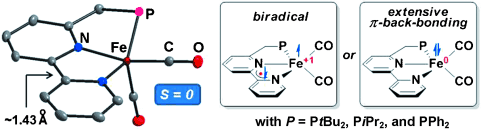
Jay J. Dunsford, Ian A. Cade, Kathlyn L. Fillman, Michael L. Neidig, and Michael J. Ingleson
Organometallics 2014, 33, 370-377
(NHC)2FeCl2 complexes undergo methoxide transfer in preference to aryl transfer from [(Aryl)B(OR)3]−, while addition of ArylBY2 (Y = Cl, OR) to (NHC)Fe–methoxide compounds leads only to formation of (NHC)BY2Aryl. The addition of PhBCl2 to (NHC)2FeX2 compounds is introduced as a method for probing NHC dissociation from iron. Two expanded ring NHCs also undergo dissociation from iron in the respective (NHC)2FeCl2 complexes.
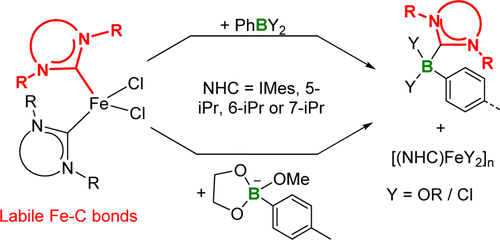
2013
Efficient Nazarov Cyclization/Wagner–Meerwein Rearrangement Terminated by a CuII‐Promoted Oxidation: Synthesis of 4‐Alkylidene Cyclopentenones
Covalency in f-element complexes
David Lebœuf, Eric Theiste, Vincent Gandon, Stephanie L. Daifuku, Michael L. Neidig, Alison J. Frontier
Chem. Eur. J. 2013, 19, 4842-4848
The discovery and elucidation of a new Nazarov cyclization/Wagner–Meerwein rearrangement/oxidation sequence is described that constitutes an efficient strategy for the synthesis of 4‐alkylidene cyclopentenones. DFT computations and EPR experiments were conducted to gain further mechanistic insight into the reaction pathways.
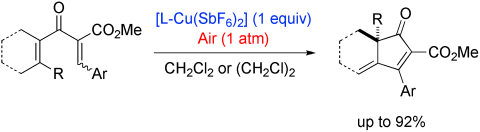
Michael L. Neidig, David L. Clark, and Richard L. Martin
Coord. Chem. Rev. 2013, 257, 394-406
The presence of covalency in complexes of the 4f and 5f elements has been a source of intense research and controversy. In addition to academic interest in this debate, there is an industrial motivation for better understanding of bonding in f-element complexes due to the need to separate trivalent trans-plutonium elements from trivalent lanthanide fission products in advanced nuclear fuel cycles. This review discusses the key evidence for covalency in f-element bonds derived from structural, spectroscopic and theoretical studies of some selected classes of molecules, including octahedral hexahalides, linear actinyl and organometallic sandwich complexes. This evidence is supplemented by a discussion of covalency, including the possibility of both overlap and near-degeneracy driven covalency and the need to quantify their relative contributions in actinide metal–ligand bonds.
Publications before Rochester
Activation of α-keto acid-dependent dioxygenases: Application of an {FeNO}7/{FeO2}8 methodology for characterizing the initial steps of O2 activation
Mechanism of the decomposition of aqueous hydrogen peroxide over hetergeneous TiSBA15 and TS-1 selective oxidation catalysts: Insights from spectroscopic and density functional theory studies
Ag K-edge EXAFS analysis of DNA-templated fluorescent silver nanoclusters: Insight into the structural origins of emission tuning by DNA sequence variations
Insight into contributions to phenol selectivity in the solution oxidation of benzene to phenol with H2O2
Direct observation of acetyl group formation from the reaction of CO with methylated H-MOR by in situ diffuse reflectance infrared spectroscopy
Formation and stabilixation of fluorescent gold nanoclusters using small molecules
The 3His triad in Dke1: Comparisons to the facial triad
Geometric structure determiation of N694C lipoxygenase: A comparative near-edge x-ray absorption spectroscopy and extended x-ray absorption fine structure study
CD and MCD of CytC3 and taurine dioxygenase: Role of the facial triad in α-KG-dependent oxygenases
Kinetic and spectroscopic studies of N694C lipoxygenase: A probe of the substrate activation mechanism of a nonheme ferric enzyme
VTVH-MCD and DFT studies of thiolate bonding in {FeNO}7/{FeNO}8 complexes of isopenicillin N synthase: Substrate determination of oxidase versus oxygenase activity in nonheme Fe enzymes
Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen-atom abstraction by non-heme iron enzymes
Spectroscopic and electronic structure studies of the role of active site interactions in the decarboxylation reaction of α-keto acid-dependent dioxygenases
Kinetic, spectroscopic, and sructural investigations of the soyvean lipoxygenases-1 first-coordiation sphere mutant, Asn684Gly
Structure-function correlation in oxygen activating non-heme iron enzymes
Spectroscopic and computatonal studies of NTBC bound to the non-heme iron enzyme (4-hydroxyphenyl)pyrucate dioxygenase: Active site contributions to drug inhibition
CD and MCD studies of the non-heme ferrous active site in (4-hydroxyphenyl)pyrucate dioxygenase: Correlation between oxygen activation in the extradiol and α-KG-dependent dioxygenase
Magnetic circular dichroism of paramagnetic species
Spectroscopic characterization of soybean lipoxygenase-1 mutants: the role of second sphere residues in the regulation of enzyme activity
Diebold, A. R.; Brown-Marshall, C. D.; Neidig, M. L.; Brownlee, J. M.; Moran, G. R.; Solomon E. I.
J. Am. Chem. Soc. 2011, 133, 18148-18160
Yoon, C. W.; Hirsekorn, K. F.; Neidig, M. L.; Yang, X.; Tilley, T.D.
ACS Catal. 2011, 1, 1665-1678
Neidig, M. L.; Sharma, J.; Yeh, H.-C.; Martinez, J. S.; Conradson, S. D.; Shreve, A. P.
J. Am. Chem. Soc. 2011, 133, 11837-11839
Neidig, M. L.; Hirsekorn, K. R.
Cat. Commun. 2011, 12, 480-484
Chen, X.; Neidig, M. L.; Tuinstra, R.; Malek, A.
J. Phys. Chem. Lett. 2010, 1, 3012-3015
Bao, Y.; Yeh, H.-C.; Zhong, C.; Sharma, J. K.; Neidig, M. L.;Cu, D. M.;Shreve, A. P.; Dyer, R. B.; Werner, J. H.; Martinez, J. S.
J. Phys. Chem. C 2010, 114, 15879-15882
Diebold, A. R.; Neidig, M. L.; Moran, G. R.; Straganz, G. D.; Solomon, E. I.
Biochemistry 2010, 49, 6945-6952
Sarangi, R.; Hocking, R. K.; Neidig, M. L.; Benfatto, M.; Holman, T. R.; Solomon, E. I.; Hodgson K. O.;Hedman, B.
Inorg. Chem. 2008, 47, 11543-11550
Neidig, M. L.; Brown, C. D.; Light, K. M.; Fujimori, D. G.; Nolan, E. M.; Price, J. C.; Barr, E. W.; Bollinger, J. M.; Krebs, C.; Solomon, E. I.
J. Am. Chem. Soc. 2007, 129, 14224-14231
Neidig, M. L.; Wecksler, A. T.; Schenk, G.; Holman, T. R.; Kacana, M.; Solomon, E. I.
J. Am. Chem. Soc. 2007, 129, 7531-7537
Brown, C. D.; Neidig, M. L.; Neibergal, M. B.; Lipscomb, J. D.; Solomon, E. I.
J. Am. Chem. Soc. 2007, 129, 7427-7438
Neidig, M. L.; Decker, A.; Choroba, O. W.; Huang, R.; Kavana, M.; Moran, G. R.; Solomon, E. I.
Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12966-12973
Neidig, M. L.; Brown, C. D.; Kavana, M.; Choroba, O. W.; Spencer, J. B.; Moran, G. R.; Solomon, E. I.
J. Inorg. Biochem. 2006, 100, 2108-2116
Segraves, E. N.; Chruszcz, M.; Neidig, M. L.; Ruddat, V.; Zhou, J.; Wecksler, A.; Minor, W.; Solomon, E. I.; Holman, T. R.
Biochemistry 2006, 45, 10233-10242
Neidig, M. L.; Solomon, E. I.
Chem. Commun. 2005, 47, 5843-5863
Neidig, M. L.; Decker, A.; Kavana, M.; Moran, G. R.; Solomon, E. I.
Biochem. Biophys. Res. Commun. 2005, 338, 206-214
Neidig, M. L.; Kavana, M.; Moran, G. R.; Solomon, E. I.
J. Am. Chem. Soc. 2004, 126, 4486-4487
Solomon, E. I.; Neidig, M. L.; Schenk, G.
Comprehensive coordination chemistry II, Elsevier/Pergamon, Amsterdam, 2004, 339-349
Schenk, G.; Neidig, M. L.; Zhou, J.; Holman, T. R.; Solomon, E. I.
Biochemistry 2003, 42, 7294-7302